School of Medicine
64 Identification of 42 Potential Colorectal Oncogenes via a RNAi Screen in Drosophila Melanogaster
Carter Niedert; Bruce Edgar (Oncological Sciences); and Peng Zhang (Oncological Sciences)
Faculty Mentor: Bruce Edgar (Oncological Sciences, University of Utah)
Abstract
Colorectal cancer is one of the most common and deadly diseases in the world. Colorectal cancer often arises from a mutated or damaged gene that leads to uncontrolled cell proliferation in the intestines. Controlling this cell proliferation by targeting the mutated or damaged genes is a potential form of cancer treatment with advantages over current treatment options. The goal of this project was to identify genes involved in damage-mediated cell proliferation which could serve as potential targets for novel cancer treatments. Using a technique known as a RNAi knockdown screen with Drosophila Melanogaster, this project found that 42 of 83 tested genes were shown to play a role in damage-mediated cell proliferation. Specifically, 14 genes were identified as promoters of cell proliferation and 28 as inhibitors. These results lay the foundation for future research that could develop methods to target these genes in colorectal cancer cells to treat patients. Such treatments could significantly improve the prognosis for colorectal cancer patients and reduce the burden of this disease on global public health.
Index Terms— Cell signaling, Gastroenterology, Genetics, Tumors, RNAi.
I. INTRODUCTION
Colorectal cancer is the third most common cancer in the United States and the second leading cause of cancer related deaths [1]. Cancer deaths occur due to the disruption of normal physiological processes caused by rapid and uncontrolled cell proliferation. Cell proliferation in cells is often regulated through cellular damage sensing pathways [2]. In the intestines, damage sensing pathways are activated by specific genes. A better understanding of the genes that are involved in intestinal damage sensing pathways could lead to possible colorectal cancer treatments that dampen these pathways and control cell proliferation.
Cell proliferation occurs rapidly in the gut due to constant exposure to stressors from the external environment [3]. Cells in the gut can sense damage caused by these stressors and trigger renewal of gut epithelial tissues via intestinal stem cell (ISC) proliferation [4]. Damaged intestinal cells can trigger ISC proliferation by releasing ligands that activate Jak-Stat signaling proliferation pathways [5]. Jak-Stat pathways mediate cytokine signaling and cytokines are commonly believed to be the main factor for activating ISC proliferation [6], [7]. Overexpression of certain Jak-Stat signaling factors has been linked to cancer development in the intestines [7]. While Jak-Stat presents an interesting target for potential cancer therapies, there are a multitude of other pathways are also involved the regulation of ISC proliferation that could be potentially targeted. For example, Wnt family signaling pathways use β-catenin as their main effector to increase or decrease ISC proliferation by activating factors such as c-Jun N-terminal kinase (JNK) or interacting with tumor suppressor genes such as APC [8], [9]. Mutations in APC and overexpression of Jak-Stat or JNK factors can lead to cancer formation and rapid cell proliferation. Due to the complexity of cellular signaling pathways, there are a staggering number of potential oncogenes that have been identified but are not yet fully understood in the context of intestinal damage-sensing. A few examples of these oncogenes include RTN1 (a type of reticulon encoding gene linked to endoplasmic reticulum function), PIEZO1 (produces a protein that links mechanical forces to biological signals), and JUND (can protect cells from p53-dependent senescence and apoptosis)[10]. These few potential oncogenes represent a much larger number of known and unknown genes that cancer cells could use to rapidly grow and evade cell death.
There are many potential cancer genes involved in damage sensing pathways and ISC proliferation that remain unidentified or poorly understood. Such genes are likely responsible for the regulation of ligand release that initially activates pathways such as Jak-Stat or Wnt [7], [9], [11]. The lack of knowledge about the function of and factors released by these potential oncogenes prevents targeted gene treatments and obscures potentially novel treatment options. An example of this is the p38 signaling pathway, which researchers have only recently discovered [12]. Researchers were able to find that damage triggers a certain signaling pathway that leads to eventual p38 activation and causes increased cell division. However, researchers were unable to understand exactly how p38 senses damage or stress and what factors it releases to promote regeneration. Understanding what factors allow p38 to sense stress could lead to physicians being able to artificially regulate the p38 pathway in cancerous cells to diminish cell growth and spread. Understanding more about other genes such as p38 could lead to discoveries about other unknown mechanisms beyond
The aim of this project is to identify which genes in the gut are responsible for sensing damage and activating cell proliferation. Disabling a gene suspected in cell proliferation pathways and quantifying the effect of its inhibition on cell proliferation is a proven method for studying gene effects. This will be done in this project by performing an RNAi gene knockdown screen using Drosophila melanogaster (i.e., fruit flies). This screen will be performed on 82 genes that have been identified through literature as being potentially involved in intestinal damage sensing pathways, such as inflammation or chemical stress pathways [14], [15]. The results of this screen could lead to a deeper understanding of damage- University of Utah UNDERGRADUATE RESEARCH JOURNAL sensing signaling pathways and to novel cancer treatments that can target specific genes. Targeted treatment of these identified genes could prevent colorectal cancer cells from rapidly proliferating and prevent them from causing the death of the patient.
II. BACKGROUND
Cancer refers to the rapid and uncontrolled growth of cells within the human body. This rapid cell growth usually arises from a mutation in a gene involved in regulating the cell cycle or programmed cell death (also known as apoptosis) [16]. Cancer cells that evade apoptosis are able to pass membranes and barriers healthy cells would not, continue undergoing cell division, and disrupt normal physiological processes [15]. This disruption can lead to death by organ failure, visceral infections or septicemia, infarction, and internal hemorrhaging [17].
Just like in healthy cells elsewhere in the body, cell division in ISCs consists of two heavily regulated stages called interphase and mitotic phase. The initiation of cell division can be triggered by several factors such as nearby cell death or damage [18]. During interphase the cell grows, replicates its DNA, and prepares to divide. The mitotic phase consists nuclear division and the physical separation of the cell into two daughter cells [19]. These daughter cells can be more ISC cells, absorptive enterocytes, goblet cells, enteroendocrine cells, or Paneth cells, each of which perform a different and necessary function in the gut [20]. In intestinal stem cells and most cells in the body, regulatory proteins known as cyclin-dependent kinases (CDK) activate at various checkpoints throughout the cycle and allow the cell to continue to subsequent stages of cell division [21]. Genes that produce or regulate the production of CDK inhibitors (CKI), such as p15, p27, p53, and p57, are able to inhibit CDKs which halts the progression of the cell cycle and prevents cell proliferation [19], [22]. CDK inhibition can often be triggered by sensed DNA damage [22]. Mutations in the genes that regulate CDK and CKI production often leads to the development of cancer as cells with DNA damage and mutations will proceed with cell division when inhibition by CKIs would otherwise prevent them from doing so [23]. Such mutations can be caused by exposure to pesticides, air pollution, metal fluids, and other chemicals [24].
Intestinal/colorectal cancer is the third most common type of cancer, likely due to the high turnover rate of the intestines and constant exposure to ingested chemicals providing more opportunity for cell mutations to occur [25]. In fact, the epithelial lining of the intestine replaces all of its cells every few days [4]. Some studies have also shown that inflammation due to illnesses such as ulcerative colitis and Crohn’s disease is another potential risk factor for developing colorectal cancer [26], [27]. Initial studies that aimed to understand cell damage and renewal in the intestines investigated the cells responsible for intestinal cell renewal, stem cells [28], [29]. Discoveries was made in 2009 and 2010 showed that specific cells called crypt stem cells were the cells-of-origin of intestinal cancer, meaning that a few mutated crypt stem cells that are able to self-renew and exhibit multipotency are responsible for the formation of large malignancies in the intestines [30], [31]. The same studies also showed the deletion of the tumor suppressor gene APC, which is also involved in Wnt-mediated proliferation pathways, led to generation of cancerous stem cells [9], [31]. Such findings led to an increased interest in understanding the pathways and genes that regulate stem cell proliferation in the intestines. A better understanding of these pathways that cancerous stem cells use to self-renew and proliferate could allow researchers and clinicians to develop treatments that target such pathways and eliminate the tumor’s self-renewing ability [32].
Chemotherapy is currently one of the most common treatments for intestinal (and many other types) of cancer [33]. Chemotherapy for colorectal cancer treatment became feasible in 1957 with the development of a compound called 5-fluorouracil (5-FU) that inhibited tumor cell division [34]. 5-FU functioned by blocking biosynthesis of essential nucleic acids in cancer cells, removing their ability to grow and divide [35]. Throughout the 1970’s and 80’s, clinical trials were performed to study the effectiveness of 5-FU treatment [36], [37]. These trials identified improved forms of treatment that generally consisted of administering 5-FU with varying amounts of a different compound (such as leucovorin). Also during the 1980’s, other treatments that inhibited epidermal growth factor were discovered and shown to be effective at limiting cancerous cell growth [38], [39]. Different compounds and chemotherapeutic treatments continued to be discovered and refined throughout the 20th and 21st century, but chemotherapy side effects such as extreme nausea, vomiting, diarrhea, long-lasting peripheral neuropathy, and severe toxicity to healthy cells make chemotherapeutic treatment a far from perfect solution [40]–[43]. This motivates the need to find an effective cancer treatment that is less harmful to the patient.
Personalized cancer treatment has become an increasingly more feasible and potentially efficacious method of treatment in recent years [44], [45]. Personalized treatment refers to determining the most effective pharmacological approach for a patient based on their own molecular characteristics [46]. Though such treatment requires the expensive process of whole-genome sequencing for each patient, its ability to identify target genes for treatment and increasing cost-effectiveness make personalized treatment more feasible each day [47], [48]. However, the complexity of cellular pathways and the many unknown genes involved in regulation of cell growth and division limit researchers’ ability to develop individualized treatments. A better understanding of cellular pathways and genetic factors would increase the feasibility of personalized cancer treatments [49], [50].
Drosophila melanogaster, also known as fruit fly, has been historically used as a powerful model to better understand physiological processes and genetic disease in humans [51]. Approximately three-fourths of all genes responsible for disease in humans have homologs in Drosophila [52]. D. melanogaster can be used as a model for the study of intestinal stem cell physiology due to the similarities of the intestinal development signaling pathways between D. melanogaster and mammals [53]. Beyond its genetic similarity to mammals, D. melanogaster is also a desirable model due to its quick life cycle and simple genetics. At 25 ℃, it takes just 9-10 days for D. melanogaster to develop from a fertilized egg to an adult and their genome only consists of four pairs of chromosomes [54]. These facts make it relatively easy and simple to breed flies with desired genes and to perform timely experiments [55].
RNAi interference is a method to perform gene knockdown which allows for the examination of any effects that a given gene may have on cell proliferation [56]. Active RNAi will cause the production of siRNA that will bind to the mRNA of a given gene. The immune system will recognize and destroy the siRNA/mRNA complex, preventing the translation of the corresponding protein and effectively silencing the targeted gene [57]. Since knocking down the function of a gene can be lethal, this method relies on the interaction between Gal80, Gal4, and UAS. Gal4 has little to no effect by itself as its main function is to bind to UAS [58], [59]. Once Gal4 has bound to UAS, the gene bound to UAS will be activated. Pairing UAS with RNAi results in the siRNA production and gene knockdown. Therefore, by crossing fly lines with Gal4 and UAS that is bound to a specific gene, the offspring will have that specific gene silenced. Gal80 is used to inhibit Gal4 at temperatures under 29 ℃ where the
flies can be crossed and maintained. Once the effects of gene knockdown are ready to be observed, placing the flies into an incubator above 29 ℃ will inhibit Gal80 and allow Gal4 to drive the previously described gene knockdown by RNAi interference [60].
RNAi screens are a popular technique for observing the effects of gene knockdown. Initial excitement for this technique was tempered by the prominence of false positives caused by off-target effects [61], [62]. However, simple solutions such as re-screening or comparing results of multiple screens through meta-analyses can verify the results of a given screen [62]. This means RNAi screens are still a convenient way to perform high-throughput experiments to identify genes of interest for a given signaling pathway or system response.
III. METHODS
A. Materials
The flies used in this project were lines of Drosophila Melanogaster with certain UAS-RNAi complexes already inserted in their genetic code. These flies were obtained from the Vienna Drosophila Resource Center in Vienna, Austria. One “driver” line of flies with a Myosin1A-Gal4 UAS-GFP tub-Gal80 complex was used in crosses with the RNAi lines to produce offspring with a complete system for gene silencing. This driver line was obtained from the Bloomington Drosophila Stock Center in Bloomington, Indiana. Myosin IA (MyoIA) was selected as a driver since it is mainly expressed in Drosophila enterocytes (mature intestinal cells) and so gene knockdown would be localized to the intestines [11]. Since UAS-GFP was included in the driver line as well, selected offspring would have both GFP fluorescence and RNAi production in the gut when shifted to 29 ℃ [63]. This allowed GFP fluorescence to serve as an indicator that the desired RNAi production and gene knockdown mechanisms were successfully incorporated into the offspring flies.
Both driver and RNAi lines were obtained with a “balancing” gene called CyO [64]. This meant that the fly lines could either pass on the driver system, UAS-RNAi complex, or the CyO gene to their offspring. Flies with two copies of the CyO gene were nonviable and quickly died. The inclusion of this balancer thus serves two purposes. First, it preserves the desired driver or RNAi system as flies breed during storage as offspring without the desired system are unable to survive or reproduce. Second, the CyO gene produces a phenotype of fly with “curly” wings. This allows visual inspection of the offspring from crosses between the driver and RNAi lines to determine if the gene knockdown system is in place (the fly has straight wings) or if one component of the knockdown system is missing (the fly has curly wings and is missing either the driver or RNAi complex).
A bacterial culture of Pseudomonas entomophilia (Pe) was used in this project to stress the intestinal cells of the flies. This culture was prepared by performing a 1:1000 dilution of Pe in Luria-Bertani (LB) broth and placing the culture in a rotating incubator at 30 ℃ and 180 rpm for 26-30 hours. The resulting culture was centrifuged at 4 ℃ at 3000rpm for 15 minutes, supernatant was discarded, and the precipitate was collected. The precipitate was mixed with a 10% sucrose in water solution to produce 400μL of “bacterial solution” for each cross that would be infected.
To make the LB broth used in the Pe culture, 950mL of water, 10g of tryptone, 10g of NaCl, and 5g of yeast extract were combined in a large glass container. The mixture was shaken and adjusted to a pH of 7.0 using sodium hydroxide. The broth was then sterilized in an autoclave for 25 minutes at 120 ℃.
Cell staining and visualization was achieved using DAPI, Alexa Fluor 488 Goat anti-Chicken antibodies, and Phospho-Histone H3 (pH3) Polyclonal antibodies obtained from Thermo Fisher Scientific, Salt Lake City, Utah. DAPI stained all present DNA for visualization of the nuclei within the gut. Alexa Fluor 488 stained enterocytes with the gene knockdown system in place and served to verify that fly crosses had been successful. pH3 allowed for visualization of cells undergoing division.
Several other different chemicals and compounds were used to prepare the fly guts for visualization. Extracted fly guts were suspended in a 1x phosphate buffered saline solution (PBS). PBS kept intestinal cells from rupturing or shriveling due to osmosis before they could be fixed. Fixation was achieved using a 4% paraformaldehyde (PFA) solution. PFA causes covalent cross-links between molecules which prevents cell decay or putrefaction. Triton X-100 diluted to 0.2% in 1x PBS (final solution called 0.2% PBST) was used to wash guts in between fixation and immunostaining steps. Triton X-100 is a nonionic surfactant that, when added to tissue that has been fixed in PFA, permeabilizes tissues prior to blocking and immunostaining. This improves the fidelity of following immunostaining. Lastly, a product called VECTASHIELD Antifade Mounting Media was placed on the final slides where guts were mounted to preserve the fluorescence of the antibodies.
Fly stocks and crosses were kept and maintained in plastic vials with about 10mL of a proprietary cornmeal-based food. Flies were kept in an 18℃ incubator when not undergoing experiments or maintenance.
B. Fly Crosses, Infection, and Dissection
For each batch performed in the screen, 10-15 virgin females of the driver line were collected and crossed with flies from one of the RNAi lines. One cross between the driver line and wildtype flies with no gene knockdown system was also done to serve as a control for each particular batch. Adult flies were removed from the vials where the crosses occurred after one week had elapsed to ensure that only pupae generated from the cross remained in each vial. Vials were kept in an 18℃ incubator for a total of about 20 days which is when the first generation of fly progeny would eclose (emerge as an adult from the pupa). Offspring with the gene knockdown system (which were identified by the lack of “curly” wings indicating desired genes were passed on) were then separated and placed in a 29 ℃ incubator to activate the knockdown system. After 5-6 days in the incubator, flies were infected with Pe by administering 200μL of Pe solution directly into the vial and by placing a paste made from the remaining Pe solution and ground yeast along the sides of the vial. Circular pieces of paper were placed in the vial beforehand to absorb the Pe culture and prevent flies from drowning in liquid. After infection, flies were left in a 29 ℃ incubator for 18 hours before being removed and dissected.
Fly guts were dissected using a stereo microscope in a dish containing 1x phosphate buffered saline (PBS). The dissection protocol was as follows: first, remove the head of the fly. Next, remove the end of the rectum which will begin pulling the gut outside of the thorax. Pull until a small amount the gut is exposed and then separate the thorax from the abdomen. This should reveal the crop, a
relatively large and white organ, which can be pulled to dislodge the gut from the abdomen. Lastly, remove the crop, slide the thorax off the gut, and remove connective tissue and organs (such as the ovaries).
C. Imaging Protocol
The dissected guts were placed into labeled centrifuge tubes and kept on ice while dissections were finished. Immediately after dissections were finished, the guts were “fixed” by adding a 16% paraformaldehyde (PFA) in PBS solution until the final solution contained 4% PFA. Guts were kept in 4% PFA solution for 25-30 minutes.
After fixation, “washes” were performed on the guts. Washing protocol consisted of removing as much liquid as possible from the tubes with a pipette and refilling the tube with a 0.2% Triton X-100 in PBS solution (0.2% PBST). Three long and three short washes were performed in between each stage of staining. Long washes consisted of placing the guts in 0.2 PBST on a rotating mixer for 15 minutes before proceeding to the next wash. Short washes consisted of removing and refilling the tube with solution without wait time. In this way, the guts in PFA were “washed” and then 10% goat serum in 0.2% PBST was added to the tubes and mixed for 25-30 minutes. This solution of goat serum acted as a blocking buffer to increase the fidelity of the subsequent immunostaining. A 1:1000 dilution of primary antibodies in PBST was then added to the tubes and the tubes were placed in a 4℃ fridge for 40-50 hours. After this time, guts were washed and a 1:1000 dilution of secondary antibodies was added to the tubes. Tubes were placed on a rotating mixer for 1.5-2 hours before guts were washed again.
Once immunostaining had been completed, guts were arranged in rows on a microscope slide with a drop of VECTASHIELD and several drops of 1x PBS. Cover slips were secured over the guts on the slides using nail polish. Slides were then labeled and ready for quantification and analysis.
D. Quantification of Dividing Cells and Data Analysis
Fluorescence microscopy was used to visualize and analyze prepared slides. pH3 positive cells were manually counted in each fly gut. Once all cells had been counted, data were input into GraphPad Prism, a data analysis software from the company Dotmatics. Unpaired T-tests were performed between the control and each RNAi line that had been prepared in that round of screening. Using the results from these tests, genes corresponding to the RNAi lines were classified as low count or high count if the p-value was less than 0.05. Low count genes, when knocked down, caused a lower amount of pH3 positive cells to be seen while high count genes increased the amount of pH3 positive cells.
IV. RESULTS
A. RNAi Screen Identified 28 High and 14 Low Count Genes
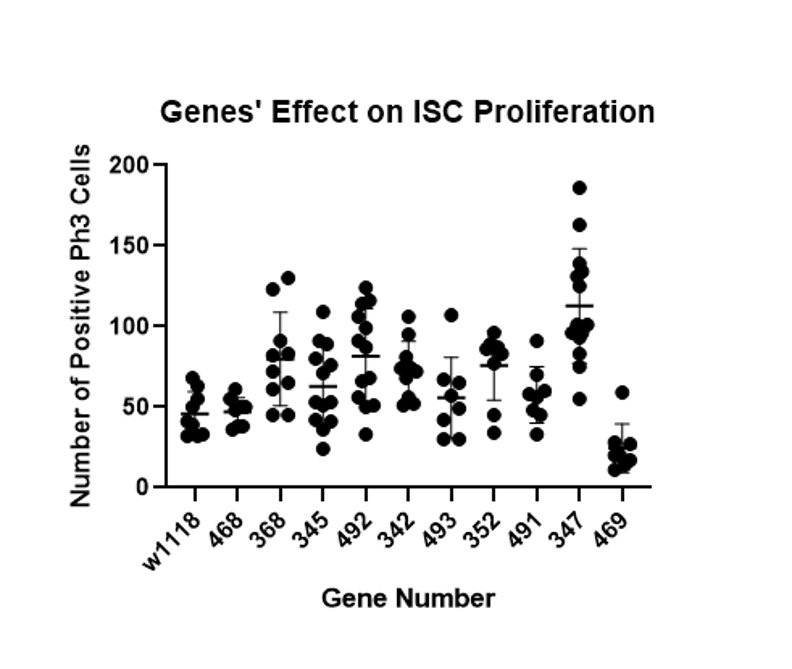
There were 82 total genes screened in this project and 42 were identified as significantly involved in ISC proliferation. As discussed in the methods section, genes were only considered significant if a t-test between the specific gene and a control comparing the amount of cell proliferation had a p-value of less than 0.05. Twelve different rounds of screening including 5-10 genes and a control were performed (see Figure 1 for an example of data collected in a batch of screening).
The several rounds of screening identified 28 genes whose knockdown led to increased cell proliferation (high count genes), 13 genes whose knockdown decreased cell proliferation
(low count genes), and 42 genes whose knockdown had no significant effect on cell proliferation. Results were
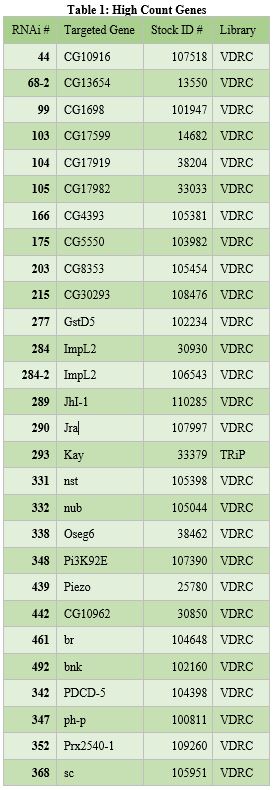
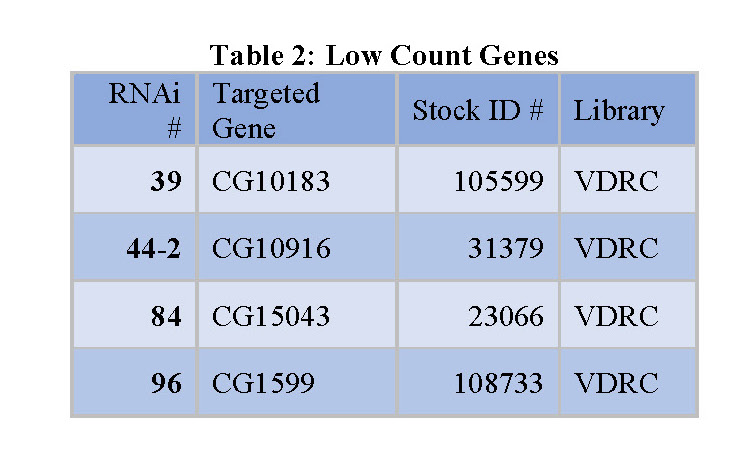
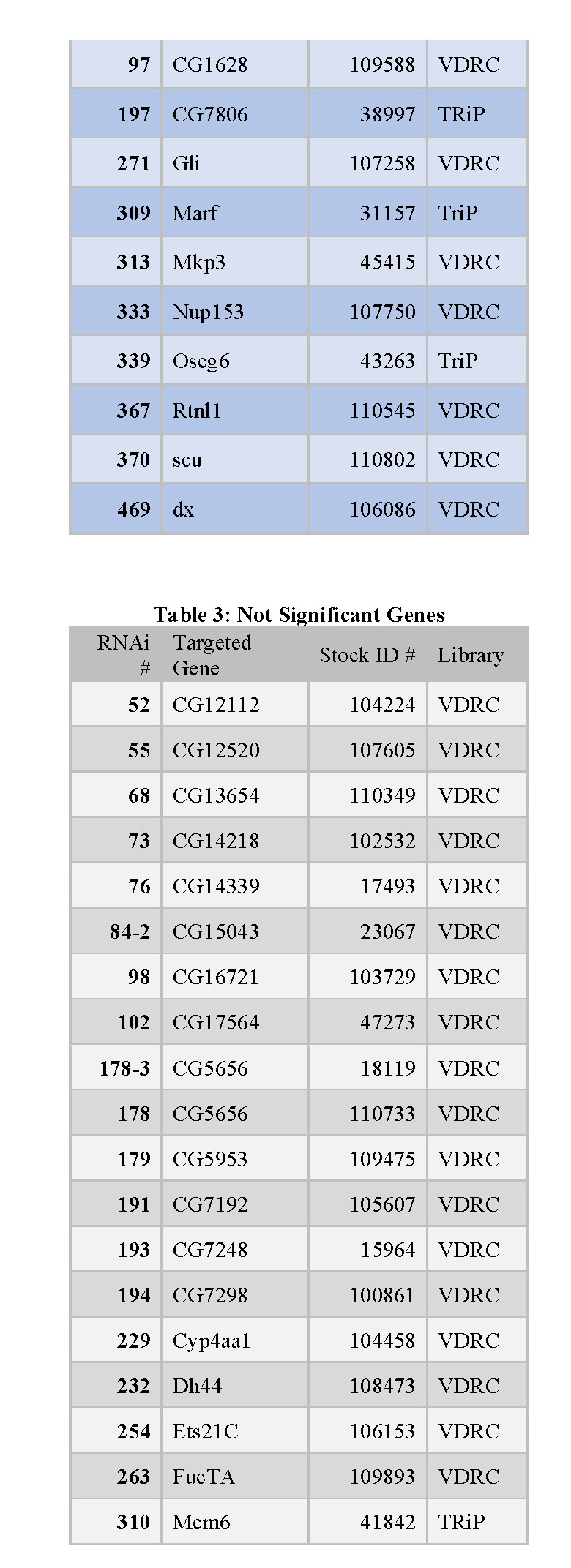
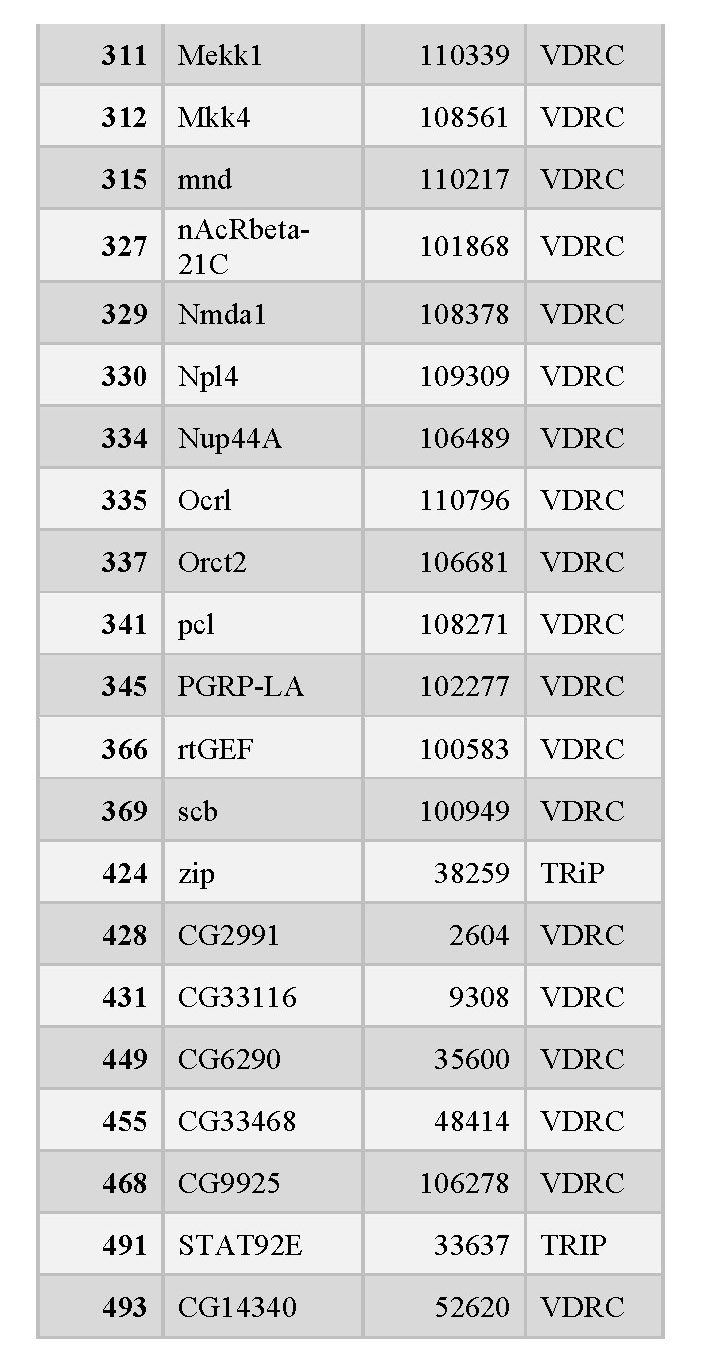
organized into tables that each contain the following information: the RNAi number used to identify fly lines within the lab, the common name of the gene targeted for knockdown, and the stock ID number for the fly library where the stock was obtained (see tables 1, 2, and 3 below).
Figure 1 – The number of dividing ISC cells were counted for each gut and RNAi line before being compared to the control (w1118) in an unpaired t-test to identify significant differences. The x-axis shows the gene number used to identify each fly line in the lab and the y-axis shows the number of dividing cells (pH3 cells). Each dot represents the dividing cell count seen in one gut for a particular gene. This batch showed gene 347 significantly increased cell proliferation and 469 decreased it (p-value<0.05).
 ‘
‘
V. DISCUSSION
Colorectal cancer is the second most common cause of cancer-related deaths in the world, with approximately 1.8 million new cases each year [1]. To better understand mechanisms of cell division and growth in colorectal cancer, this project aimed to identify which genes in the gut are responsible for sensing damage and activating cell proliferation. Disabling a gene suspected in cell proliferation pathways and quantifying the effect on cell proliferation is a proven method for studying genes. In this project, genes were disabled and studied by performing an RNAi gene knockdown screen using Drosophila melanogaster (i.e., fruit flies). Of the 82 genes that went through the screen, 42 were identified as significant in regulating cell proliferation. To inhibit colorectal cancer cell’s ability to grow and spread, future treatments could potentially target any of the 42 identified genes. Though more work is necessary to develop such treatments, these results lay the foundation for more effective treatments that will reduce the worldwide impact of colorectal cancer.
Knockdown or silencing of the 42 identified genes in the screen caused cell proliferation to either increase or decrease (see Figure 1). Since the fly intestines were infected with bacteria and damaged as part of the screening process, it is likely that the 42 identified genes play some role in regulating damage-mediated cell proliferation. The 14 “low count genes” likely promote or upregulate cell proliferation in response to damage (see Table 2). The 28 “high count genes” likely do the opposite; inhibit or downregulate damage-mediated cell proliferation (see Table 1).
Many of the potential colorectal oncogenes identified in this project have orthologs in humans that could potentially be targeted for cancer treatment. For example, For example, the “Marf” gene (see Table 2) has a DIOPT score of 13 out of 19 with the human gene MFN2 [65], [66]. This score corresponds to the level of consensus among gene databases and scientific papers that the two genes are indeed orthologs. This relatively high score indicates that MFN2 is possibly involved in intestinal damage-sensing and cell proliferation pathways as Marf is in flies. MFN2 has already shown to be involved in cell proliferation elsewhere in the body, which supports that its identification as a potentially significant gene in intestinal cell renewal was likely accurate [67]. Many other genes identified in this screen have DIOPT scores as high as 15,
with several of human orthologs of genes such as “scu” and “Oseg6” (see Table 2) not having previously been implicated in cell proliferation pathways. Any one of these orthologs could be furthered studied to lead to potential new colorectal cancer treatments.
The two main limitations of this screening project were the inability implement techniques for mitigating off-target effects and sample sizes of five or less for certain genes. It is known that RNAi screens can have false positives due to off-target effects of the siRNAs where genes other than the targeted one are silenced or affected [68]. Though several techniques exist to mitigate such off-target effects, they were could not feasibly be implemented in this project [69]. Second, due to rare instances of fly mishandling or premature fly death, some lines were screen with relatively small sample sizes (<5).
Despite project limitations, it is still likely that the 42 identified genes could serve as potential cancer therapy targets. For example, look at the case of the long-studied ADAR1 gene. Though previously only implicated as a potential target for cancer treatment, recent studies that better elucidated its mechanisms have allowed researchers to begin formulating ideas for new cancer treatments that would target this gene [70]. Though it is possible that many of the 42 identified are not feasible targets, these results allow researchers to focus their efforts on a smaller pool of potential targets rather than making educated guesses on which genes to start with. This could save large amounts of time and money from being spent on fruitless projects, which can then be reallocated to more promising ones. Ideally, this means that better cancer treatments are discovered quicker and made available to patients sooner.
Future work for this project entails performing a secondary screen on the “high count genes” and possibly re-screening genes that had low sample sizes. The secondary screen on the high-count genes will be performed without a bacterial infection. If knockdown of one of these genes still results in increased cell proliferation despite the intestine not facing damage or stress due to bacterial toxins, it is likely those genes are not involved in damage-mediated cell proliferation. However, if knockdown of a given high-count gene without infection shows similar cell division to that of the control, then it would be very likely that the gene does play a role in damage-mediated cell proliferation. Future studies are being considered to explore the function of the genes that caused the greatest changes in cell proliferation, but are still in planning stages at this moment.
Colorectal cancer continues to be one of the world’s most prevalent and deadly diseases [1]. Recent studies seem to suggest that its impact on younger and younger generations will increase in the coming years [71], [72]. Current treatment options for colorectal cancer, such as chemotherapy and surgery, are less than optimal as they often harm healthy cells as well as cancer cells [73], [74]. Though the results of this screening project do not immediately benefit colorectal cancer patients, they do lay the foundation for future studies and improved cancer therapies that could vastly improve colorectal cancer treatment options. These optimized treatments could be more affordable, less damaging to healthy cells, and lead to improved patient outcomes. Such treatments would dramatically reduce the global financial burden caused by colorectal cancer and, most importantly, would help to reduce the nearly 1 million deaths that it causes each year [1].
REFERENCES
[1] H. Sung et al., “Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries,” CA. Cancer J. Clin., vol. 71, no. 3, pp. 209–249, May 2021, doi: 10.3322/caac.21660.
[2] R. J. Duronio and Y. Xiong, “Signaling Pathways that Control Cell Proliferation,” Cold Spring Harb. Perspect. Biol., vol. 5, no. 3, pp. a008904–a008904, Mar. 2013, doi: 10.1101/cshperspect.a008904.
[3] W. M. Wong and N. A. Wright, “Cell proliferation in gastrointestinal mucosa,” J. Clin. Pathol., vol. 52, no. 5, pp. 321–333, May 1999, doi: 10.1136/jcp.52.5.321.
[4] C. Crosnier, D. Stamataki, and J. Lewis, “Organizing cell renewal in the intestine: stem cells, signals and combinatorial control,” Nat. Rev. Genet., vol. 7, no. 5, pp. 349–359, May 2006, doi: 10.1038/nrg1840.
[5] B. K. Staley and K. D. Irvine, “Warts and Yorkie Mediate Intestinal Regeneration by Influencing Stem Cell Proliferation,” Curr. Biol., vol. 20, no. 17, pp. 1580–1587, Sep. 2010, doi: 10.1016/j.cub.2010.07.041.
[6] C. Andrews, M. H. McLean, and S. K. Durum, “Cytokine Tuning of Intestinal Epithelial Function,” Front. Immunol., vol. 9, p. 1270, Jun. 2018, doi: 10.3389/fimmu.2018.01270.
[7] P. Sansone and J. Bromberg, “Targeting the Interleukin-6/Jak/Stat Pathway in Human Malignancies,” J. Clin. Oncol., vol. 30, no. 9, pp. 1005–1014, Mar. 2012, doi: 10.1200/JCO.2010.31.8907.
[8] A. Casali and E. Batlle, “Intestinal Stem Cells in Mammals and Drosophila,” Cell Stem Cell, vol. 4, no. 2, pp. 124–127, Feb. 2009, doi: 10.1016/j.stem.2009.01.009.
[9] M. Krausova and V. Korinek, “Wnt signaling in adult intestinal stem cells and cancer,” Cell. Signal., vol. 26, no. 3, pp. 570–579, Mar. 2014, doi: 10.1016/j.cellsig.2013.11.032.
[10] E. W. Sayers et al., “Database resources of the national center for biotechnology information,” Nucleic Acids Res., vol. 50, no. D1, pp. D20–D26, Jan. 2022, doi: 10.1093/nar/gkab1112.
[11] H. Jiang, P. H. Patel, A. Kohlmaier, M. O. Grenley, D. G. McEwen, and B. A. Edgar, “Cytokine/Jak/Stat Signaling Mediates Regeneration and Homeostasis in the Drosophila Midgut,” Cell, vol. 137, no. 7, pp. 1343–1355, Jun. 2009, doi: 10.1016/j.cell.2009.05.014.
[12] P. H. Patel et al., “Damage sensing by a Nox-Ask1-MKK3-p38 signaling pathway mediates regeneration in the adult Drosophila midgut,” Nat. Commun., vol. 10, no. 1, p. 4365, Dec. 2019, doi: 10.1038/s41467-019-12336-w.
[13] H. Jiang, A. Tian, and J. Jiang, “Intestinal stem cell response to injury: lessons from Drosophila,” Cell. Mol. Life Sci., vol. 73, no. 17, pp. 3337–3349, Sep. 2016, doi: 10.1007/s00018-016-2235-9.
[14] T. Jess et al., “Risk of Intestinal Cancer in Inflammatory Bowel Disease: A Population-Based Study From Olmsted County, Minnesota,” Gastroenterology, vol. 130, no. 4, pp. 1039–1046, Apr. 2006, doi: 10.1053/j.gastro.2005.12.037.
[15] D. R. Green, “Cell Death and Cancer,” Cold Spring Harb. Perspect. Biol., vol. 14, no. 9, p. a041103, Sep. 2022, doi:
10.1101/cshperspect.a041103.
[16] J. C. Reed, “Mechanisms of Apoptosis,” Am. J. Pathol., vol. 157, no. 5, pp. 1415–1430, Nov. 2000, doi: 10.1016/S0002-9440(10)64779-7.
[17] J. Inagaki, V. Rodriguez, and G. P. Bodey, “Causes of death in cancer patients,” Cancer, vol. 33, no. 2, pp. 568–573, Feb. 1974, doi: 10.1002/1097-0142(197402)33:2<568::AID-CNCR2820330236>3.0.CO;2-2.
[18] C. Franceschi, “Cell proliferation, cell death and aging,” Aging Clin. Exp. Res., vol. 1, no. 1, pp. 3–15, Sep. 1989, doi: 10.1007/BF03323871.
[19] K. Vermeulen, D. R. Van Bockstaele, and Z. N. Berneman, “The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer: Cell cycle regulation and deregulation,” Cell Prolif., vol. 36, no. 3, pp. 131–149, Jun. 2003, doi: 10.1046/j.1365-2184.2003.00266.x.
[20] T. K. Noah and N. F. Shroyer, “Notch in the Intestine: Regulation of Homeostasis and Pathogenesis,” Annu. Rev. Physiol., vol. 75, no. 1, pp. 263–288, Feb. 2013, doi: 10.1146/annurev-physiol-030212-183741.
[21] J. Y. Ong and J. Z. Torres, “Dissecting the mechanisms of cell division,” J. Biol. Chem., vol. 294, no. 30, pp. 11382–11390, Jul. 2019, doi: 10.1074/jbc.AW119.008149.
[22] L. Ding et al., “The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer,” Int. J. Mol. Sci., vol. 21, no. 6, p. 1960, Mar. 2020, doi: 10.3390/ijms21061960.
[23] J. Wade Harper and S. J. Elledge, “Cdk inhibitors in development and cancer,” Curr. Opin. Genet. Dev., vol. 6, no. 1, pp. 56–64, Feb. 1996, doi: 10.1016/S0959-437X(96)90011-8.
[24] R. W. Clapp, M. M. Jacobs, and E. L. Loechler, “Environmental and Occupational Causes of Cancer: New Evidence 2005-2007,” Rev. Environ. Health, vol. 23, no. 1, pp. 1–38, Jan. 2008, doi: 10.1515/REVEH.2008.23.1.1.
[25] E. J. Davies, V. Marsh, and A. R. Clarke, “Origin and maintenance of the intestinal cancer stem cell: ORIGIN AND MAINTENANCE OF THE ICSC,” Mol. Carcinog., vol. 50, no. 4, pp. 254–263, Apr. 2011, doi: 10.1002/mc.20631.
[26] O. Kiraly, G. Gong, W. Olipitz, S. Muthupalani, and B. P. Engelward, “Inflammation-Induced Cell Proliferation Potentiates DNA Damage-Induced Mutations In Vivo,” PLOS Genet., vol. 11, no. 2, p. e1004901, Feb. 2015, doi: 10.1371/journal.pgen.1004901.
[27] T. A. Ullman and S. H. Itzkowitz, “Intestinal Inflammation and Cancer,” Gastroenterology, vol. 140, no. 6, pp. 1807-1816.e1, May 2011, doi: 10.1053/j.gastro.2011.01.057.
[28] F. Radtke and H. Clevers, “Self-Renewal and Cancer of the Gut: Two Sides of a Coin,” Science, vol. 307, no. 5717, pp. 1904–1909, Mar. 2005, doi: 10.1126/science.1104815.
[29] F. Radtke, H. Clevers, and O. Riccio, “From Gut Homeostasis to Cancer,” Curr. Mol. Med., vol. 6, no. 3, pp. 275–289, May 2006, doi: 10.2174/156652406776894527.
[30] N. Barker et al., “Crypt stem cells as the cells-of-origin of intestinal cancer,” Nature, vol. 457, no. 7229, pp. 608–611, Jan. 2009, doi: 10.1038/nature07602.
[31] F. J. Abdul Khalek, G. I. Gallicano, and L. Mishra, “Colon cancer stem cells,” Gastrointest. Cancer Res. GCR, no. Suppl 1, pp. S16-23, Nov. 2010.
[32] K. Chen, Y. Huang, and J. Chen, “Understanding and targeting cancer stem cells: therapeutic implications and challenges,” Acta Pharmacol. Sin., vol. 34, no. 6, pp. 732–740, Jun. 2013, doi: 10.1038/aps.2013.27.
[33] M. M. Olsen, K. B. LeFebvre, K. J. Brassil, and Oncology Nursing Society, Eds., Chemotherapy and immunotherapy guidelines and recommendations for practice. Pittsburgh, Pennsylvania: Oncology Nursing Society, 2019.
[34] B. Gustavsson et al., “A Review of the Evolution of Systemic Chemotherapy in the Management of Colorectal Cancer,” Clin. Colorectal Cancer, vol. 14, no. 1, pp. 1–10, Mar. 2015, doi: 10.1016/j.clcc.2014.11.002.
[35] D. B. Longley, D. P. Harkin, and P. G. Johnston, “5-Fluorouracil: mechanisms of action and clinical strategies,” Nat. Rev. Cancer, vol. 3, no. 5, pp. 330–338, May 2003, doi: 10.1038/nrc1074.
[36] N. Petrelli et al., “A prospective randomized trial of 5-fluorouracil versus 5-fluorouracil and high-dose leucovorin versus 5-fluorouracil and methotrexate in previously untreated patients with advanced colorectal carcinoma.,” J. Clin. Oncol., vol. 5, no. 10, pp. 1559–1565, Oct. 1987, doi: 10.1200/JCO.1987.5.10.1559.
[37] S. Waxman and H. Bruckner, “The enhancement of 5-fluorouracil antimetabolic activity by leucovorin, menadione and α-tocopherol,” Eur. J. Cancer Clin. Oncol., vol. 18, no. 7, pp. 685–692, Jul. 1982, doi: 10.1016/0277-5379(82)90215-2.
[38] C. M. Stoscheck and L. E. King Jr., “Role of Epidermal Growth Factor in Carcinogenesis,” Cancer Res., vol. 46, no. 3, pp. 1030–1037, Mar. 1986.
[39] Y. Imai, C. K. H. Leung, H. G. Friesen, and R. P. C. Shiu, “Epidermal Growth Factor Receptors and Effect of Epidermal Growth Factor on Growth of Human Breast Cancer Cells in Long-Term Tissue Culture1,” Cancer Res., vol. 42, no. 11, pp. 4394–4398, Nov. 1982.
[40] C. Tofthagen, “Surviving Chemotherapy for Colon Cancer and Living with the Consequences,” J. Palliat. Med., vol. 13, no. 11, pp. 1389–1391, Nov. 2010, doi: 10.1089/jpm.2010.0124.
[41] E. P. M. de Almeida, M. G. R. de Gutiérrez, and N. P. Adami, “Monitoramento e avaliação dos efeitos colaterais da quimioterapia em pacientes com câncer de cólon,” Rev. Lat. Am. Enfermagem, vol. 12, no. 5, pp. 760–766, Oct. 2004, doi: 10.1590/S0104-11692004000500009.
[42] QUASAR Collaborative Group, “Adjuvant chemotherapy versus observation in patients with colorectal cancer: a
randomised study,” The Lancet, vol. 370, no. 9604, pp. 2020–2029, Dec. 2007, doi: 10.1016/S0140-6736(07)61866-2.
[43] P. M. Wigmore, S. Mustafa, M. El-Beltagy, L. Lyons, J. Umka, and G. Bennett, “Effects of 5-FU,” in Chemo Fog, vol. 678, R. B. Raffa and R. J. Tallarida, Eds. New York, NY: Springer New York, 2010, pp. 157–164. doi: 10.1007/978-1-4419-6306-2_20.
[44] T. Lan, H. Que, M. Luo, X. Zhao, and X. Wei, “Genome editing via non-viral delivery platforms: current progress in personalized cancer therapy,” Mol. Cancer, vol. 21, no. 1, p. 71, Mar. 2022, doi: 10.1186/s12943-022-01550-8.
[45] X. Lai et al., “A scalable solver for a stochastic, hybrid cellular automaton model of personalized breast cancer therapy,” Int. J. Numer. Methods Biomed. Eng., vol. 38, no. 1, Jan. 2022, doi: 10.1002/cnm.3542.
[46] C. Rodríguez-Antona and M. Taron, “Pharmacogenomic biomarkers for personalized cancer treatment,” J. Intern. Med., vol. 277, no. 2, pp. 201–217, Feb. 2015, doi: 10.1111/joim.12321.
[47] K. G. Samsom et al., “Study protocol: Whole genome sequencing Implementation in standard Diagnostics for Every cancer patient (WIDE),” BMC Med. Genomics, vol. 13, no. 1, p. 169, Dec. 2020, doi: 10.1186/s12920-020-00814-w.
[48] K. Schwarze, J. Buchanan, J. C. Taylor, and S. Wordsworth, “Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature,” Genet. Med., vol. 20, no. 10, pp. 1122–1130, Oct. 2018, doi: 10.1038/gim.2017.247.
[49] C. S. Shemesh et al., “Personalized Cancer Vaccines: Clinical Landscape, Challenges, and Opportunities,” Mol. Ther., vol. 29, no. 2, pp. 555–570, Feb. 2021, doi: 10.1016/j.ymthe.2020.09.038.
[50] A. Burguin, C. Diorio, and F. Durocher, “Breast Cancer Treatments: Updates and New Challenges,” J. Pers. Med., vol. 11, no. 8, p. 808, Aug. 2021, doi: 10.3390/jpm11080808.
[51] Z. Mirzoyan, M. Sollazzo, M. Allocca, A. M. Valenza, D. Grifoni, and P. Bellosta, “Drosophila melanogaster: A Model Organism to Study Cancer,” Front. Genet., vol. 10, p. 51, Mar. 2019, doi: 10.3389/fgene.2019.00051.
[52] B. Ugur, K. Chen, and H. J. Bellen, “Drosophila tools and assays for the study of human diseases,” Dis. Model. Mech., vol. 9, no. 3, pp. 235–244, Mar. 2016, doi: 10.1242/dmm.023762.
[53] Y. Apidianakis and L. G. Rahme, “Drosophila melanogaster as a model for human intestinal infection and pathology,” Dis. Model. Mech., vol. 4, no. 1, pp. 21–30, Jan. 2011, doi: 10.1242/dmm.003970.
[54] M. Yamaguchi and H. Yoshida, “Drosophila as a Model Organism,” in Drosophila Models for Human Diseases, vol. 1076, M. Yamaguchi, Ed. Singapore: Springer Singapore, 2018, pp. 1–10. doi: 10.1007/978-981-13-0529-0_1.
[55] K. G. Hales, C. A. Korey, A. M. Larracuente, and D. M. Roberts, “Genetics on the Fly: A Primer on the Drosophila Model System,” Genetics, vol. 201, no. 3, pp. 815–842, Nov. 2015, doi: 10.1534/genetics.115.183392.
[56] R. C. Wilson and J. A. Doudna, “Molecular Mechanisms of RNA Interference,” Annu. Rev. Biophys., vol. 42, no. 1, pp. 217–239, May 2013, doi: 10.1146/annurev-biophys-083012-130404.
[57] N. Perrimon, J.-Q. Ni, and L. Perkins, “In vivo RNAi: Today and Tomorrow,” Cold Spring Harb. Perspect. Biol., vol. 2, no. 8, pp. a003640–a003640, Aug. 2010, doi: 10.1101/cshperspect.a003640.
[58] M. L. Suster, L. Seugnet, M. Bate, and M. B. Sokolowski, “Refining GAL4-driven transgene expression inDrosophila with a GAL80 enhancer-trap,” genesis, vol. 39, no. 4, pp. 240–245, Aug. 2004, doi: 10.1002/gene.20051.
[59] A. H. Brand and N. Perrimon, “Targeted gene expression as a means of altering cell fates and generating dominant phenotypes,” Development, vol. 118, no. 2, pp. 401–415, Jun. 1993, doi: 10.1242/dev.118.2.401.
[60] S. E. McGuire, P. T. Le, A. J. Osborn, K. Matsumoto, and R. L. Davis, “Spatiotemporal Rescue of Memory Dysfunction in Drosophila,” Science, vol. 302, no. 5651, pp. 1765–1768, Dec. 2003, doi: 10.1126/science.1089035.
[61] F. D. Sigoillot and R. W. King, “Vigilance and Validation: Keys to Success in RNAi Screening,” ACS Chem. Biol., vol. 6, no. 1, pp. 47–60, Jan. 2011, doi: 10.1021/cb100358f.
[62] S. E. Mohr, J. A. Smith, C. E. Shamu, R. A. Neumüller, and N. Perrimon, “RNAi screening comes of age: improved techniques and complementary approaches,” Nat. Rev. Mol. Cell Biol., vol. 15, no. 9, pp. 591–600, Sep. 2014, doi: 10.1038/nrm3860.
[63] Z. Zhai et al., “Accumulation of differentiating intestinal stem cell progenies drives tumorigenesis,” Nat. Commun., vol. 6, no. 1, p. 10219, Dec. 2015, doi: 10.1038/ncomms10219.
[64] D. E. Miller, K. R. Cook, and R. S. Hawley, “The joy of balancers,” PLOS Genet., vol. 15, no. 11, p. e1008421, Nov. 2019, doi: 10.1371/journal.pgen.1008421.
[65] K. Clark, I. Karsch-Mizrachi, D. J. Lipman, J. Ostell, and E. W. Sayers, “GenBank,” Nucleic Acids Res., vol. 44, no. D1, pp. D67–D72, Jan. 2016, doi: 10.1093/nar/gkv1276.
[66] Y. Hu et al., “FlyRNAi.org—the database of the Drosophila RNAi screening center and transgenic RNAi project: 2021 update,” Nucleic Acids Res., vol. 49, no. D1, pp. D908–D915, Jan. 2021, doi: 10.1093/nar/gkaa936.
[67] P. Zanfardino and V. Petruzzella, “Autophagy and proliferation are dysregulated in Charcot-Marie-Tooth disease type 2A cells harboring MFN2 (mitofusin 2) mutation,” Autophagy Rep., vol. 1, no. 1, pp. 537–541, Dec. 2022, doi: 10.1080/27694127.2022.2132447.
[68] N. Schultz et al., “Off-target effects dominate a large-scale RNAi screen for modulators of the TGF-β pathway and reveal microRNA regulation of TGFBR2,” Silence, vol. 2, no. 1, p. 3, 2011, doi: 10.1186/1758-907X-2-3.
[69] A. L. Jackson and P. S. Linsley, “Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application,” Nat. Rev. Drug Discov., vol. 9, no. 1, pp. 57–67, Jan. 2010, doi: 10.1038/nrd3010.
[70] A. R. Baker and F. J. Slack, “ADAR1 and its implications in cancer development and treatment,” Trends Genet., vol. 38, no.
8, pp. 821–830, Aug. 2022, doi: 10.1016/j.tig.2022.03.013.
[71] E. Feletto et al., “Trends in Colon and Rectal Cancer Incidence in Australia from 1982 to 2014: Analysis of Data on Over 375,000 Cases,” Cancer Epidemiol. Biomarkers Prev., vol. 28, no. 1, pp. 83–90, Jan. 2019, doi: 10.1158/1055-9965.EPI-18-0523.
[72] R. L. Siegel, C. D. Jakubowski, S. A. Fedewa, A. Davis, and N. S. Azad, “Colorectal Cancer in the Young: Epidemiology, Prevention, Management,” Am. Soc. Clin. Oncol. Educ. Book, no. 40, pp. e75–e88, May 2020, doi: 10.1200/EDBK_279901.
[73] S. M. Koroukian, F. Xu, P. M. Bakaki, M. Diaz-Insua, T. P. Towe, and C. Owusu, “Comorbidities, Functional Limitations, and Geriatric Syndromes in Relation to Treatment and Survival Patterns Among Elders With Colorectal Cancer,” J. Gerontol. A. Biol. Sci. Med. Sci., vol. 65A, no. 3, pp. 322–329, Mar. 2010, doi: 10.1093/gerona/glp180.
[74] D. Symeonidis, G. Christodoulidis, G. Koukoulis, M. Spyridakis, and K. Tepetes, “Colorectal cancer surgery in the elderly: limitations and drawbacks,” Tech. Coloproctology, vol. 15, no. S1, pp. 47–50, Oct. 2011, doi: 10.1007/s10151-011-0751-z.
