School of Medicine
57 Biochemical analysis of the Smoothened / Protein Kinase A binding interaction in Hedgehog signaling
Nathan Iverson
Faculty Mentor: Benjamin Myers (Oncological Sciences, University of Utah)
Abstract
PKA-C complexes is needed to answer these questions, because SMO is the first documented GPCR to directly sequester PKA-C at the membrane, and the first example of a PKA-C decoy substrate whose binding is regulated by phosphorylation. Here I use several in vitro biochemical and structural approaches to address this key knowledge gap. First, I attempted to directly test the role of GRK2/3 phosphorylation in regulating the SMO/PKA-C interaction using binding assays between PKA-C and SMOct engineered to contain mutations that mimic the GRK2/3 phosphorylation events. Second, I prepared a crosslinked complex of SMOct/PKA-C for crystallography studies, thereby overcoming the intrinsically low affinity binding interaction between SMOct and PKA-C that has hampered structural characterization of the complex. Finally, I optimized a protocol for isotopically labeled SMOct preparation in E. coli to be used in future NMR studies to overcome the challenges presented by characterizing this largely unstructured domain via traditional crystallography or cryoEM approaches. Understanding the structural and biochemical basis for the SMO-PKA complex will provide critical insights into an essential step in Hh signal transduction and may lead to more effective therapeutic agents to treat a range of different cancers.
INTRODUCTION
The Hedgehog signaling pathway is an essential player in vertebrate embryonic organ development, controlling the formation of nearly every organ in our bodies. This signaling pathway was initially discovered by researchers Nusslein-Volhard and Wieschaus in 1980 from their work in Drosophila melanogaster development. The Hh pathway received its name due to the spikey, hedgehog-like appearance of the fruit fly larvae when the Hh gene is knocked out1. Cell proliferation and tissue patterning during development are tuned through Hh ligand gradients that allow for regulated pathway activity in a dose-dependent manner. This promotes cells to adopt specific fates allowing for complex developmental processes and patterning2. The pathway also has roles in tissue homeostasis and cell maintenance postnatally. Insufficient Hh pathway activity during development results in birth defects or embryonic death. Improper activation postnatally is associated with many cancers including basal cell carcinoma and medulloblastoma. Despite the pathway’s importance and association with disease, the signal transduction mechanism has largely remained a mystery.
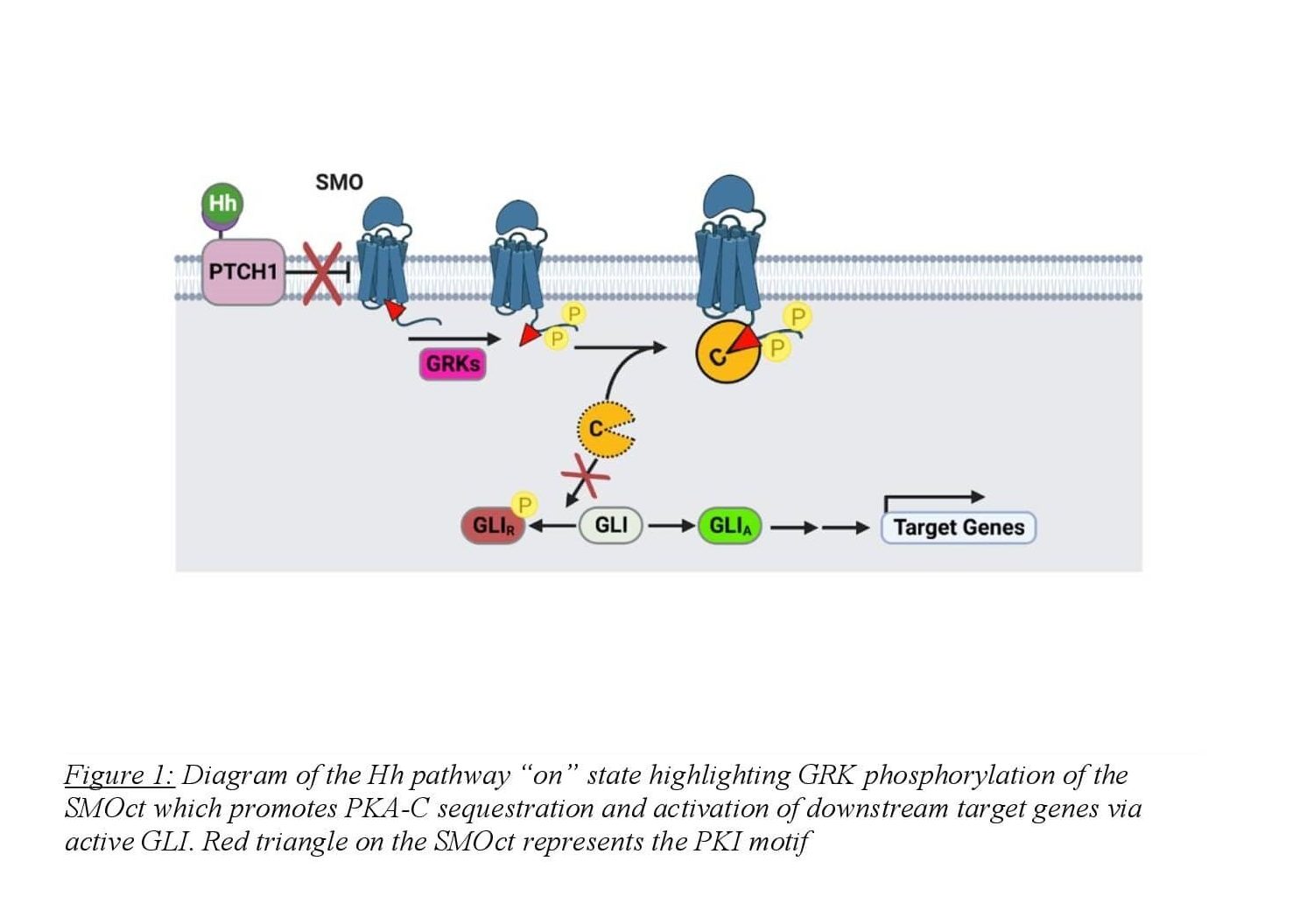
SMO is a seven-pass transmembrane protein that is the principal determinant of Hh signal transduction at the cell surface. In the pathway “off” state, SMO is inhibited by the transmembrane protein Patched 1 (PTCH1) through an indirect mechanism3. PTCH1 inhibits pathway activity through the transport of cholesterol, which is an endogenous SMO activating ligand4. Researchers have found that PTCH1 depletes cholesterol from the inner leaflet of the plasma membrane rendering the sterol inaccessible to SMO4. This harboring of cholesterol at the outer leaflet of the membrane inhibits SMO activation and expression of target genes.
In the pathway “on” state, Hh ligand binds to PTCH1 which abolishes the transport function of the transmembrane protein, allowing equilibration of cholesterol between the inner and outer leaflet of the plasma membrane. Recent research shows that SMO is activated by a membrane sterol entering a hydrophobic tunnel that connects the inner leaflet to a site in the seven-transmembrane domain of SMO5. The binding of cholesterol induces a conformational change to the protein, initiating the active state of SMO and signaling cascade that leads to the expression of target genes.
Active state SMO initiates activation of the transcription factor GLI through a series of events that was poorly understood until recently. PKA-C is known to be the main regulator of GLI in the Hh pathway6. In the pathway “off” state, PKA is unrestrained and phosphorylates GLI targeting it by a ubiquitinating complex and degraded by the proteasome. When GLI is not phosphorylated by PKA-C, it is an activated transcription factor to promote expression of target genes. We hypothesize that PKA-C activity in the Hh pathway is regulated through direct inhibition by SMO. PKA-C activity is commonly regulated by signal transduction from GPCRs. In canonical GPCR signal transduction, active GPCRs bind to heterotrimeric G proteins at a cytoplasmic domain allowing GTP-bound Gα and free Gβγ to transduce the signal to downstream effector proteins. Typically, GPCRs inhibit PKA-C via coupling to inhibitory heterotrimeric G proteins, which trigger reduction in cytoplasmic levels of cAMP through the membrane protein adenylyl cyclase. cAMP binds to PKA regulatory proteins releasing them from inhibiting PKA-C, and allowing phosphorylation of downstream targets, such as GLI. SMO, however, does not appear to utilize this canonical mechanism to inactivate PKA-C, as both genetic or pharmacological strategies to block inhibitory G proteins do not alter GLI transcriptional activity or expression of Hh pathway target genes7,8. Thus, another pathway mechanism was needed to explain Hh signal transduction.
Arveseth and colleagues uncovered this unconventional mechanism in 2021 finding that SMO directly binds to PKA-C in the active state, harboring the kinase at the membrane and inhibiting its enzymatic activity8,9 (Figure 1). SMO binds to PKA-C via a pseudosubstrate domain present on the cytoplasmic tail of SMO with an amino acid sequence similar to protein kinase inhibitor (PKI)9. PKI is a small, largely unstructured, cytoplasmic protein that binds to the active site of PKA-C with nanomolar affinity. The inhibition of PKA-C is due to PKI’s N-terminal pseudosubstrate domain. This domain binds to the active site of the enzyme independently of cAMP levels and is unable to be phosphorylated, making PKI a potent competitive inhibitor of PKA-C10. The researchers found that SMOct contains a PKI motif in the proximal domain of the tail with four hallmarks that resemble known PKA-C pseudosubstrates. First, the SMOct contains the nonphosphorylatable residue alanine at the canonical phosphorylation site (P site) for PKA-C substrates. The SMOct also contains arginines at the P-2 and P-3 positions that have been shown to be necessary for other pseudosubstrates such as PKI. Finally, SMOct contains a hydrophobic residue at the P+1 site as well as a predicted α-helical domain N-terminal to the pseudosubstrate domain9. These four characteristics are essential in high-affinity binding between PKA-C and pseudosubstrates. These findings suggest a mechanism for how SMOct binds and inhibits PKA-C enzymatic activity at the membrane.
In the pathway “on” state, activated SMO undergoes phosphorylation of the cytosolic tail by GRK2/3 which initiates PKA-C binding. The SMOct/PKA-C binding interaction is likely enhanced when SMOct is phosphorylated by GRK2/3, though this has never been shown directly. Predicted models of the SMOct/PKA-C binding complex suggest a crucial role for phosphorylation of the SMOct in promoting a conformation that will bind to PKA-C. The SMOct/PKA-C binding interaction is relatively weak with an equilibrium dissociation constant (KD) of 752 nM9. This differs greatly from the PKI/PKA-C binding interaction which has a KD of 1.1 nM11. Seven GRK2/3 phosphorylation sites present in 3 clusters have been identified on the proximal domain of the SMOct using mass spectrometry8. These sites exhibit dependence on SMO’s activity state and mediate the binding of PKA-C. In addition, two phosphorylation sites are also present on the proximal side of SMOct that are GRK2/3 independent and are weakly associated with SMO activity. Conservation of these 9 phosphorylation sites is seen across many vertebrates suggesting the importance of their role in Hh signaling. When these residues on SMOct involved in GRK2/3 phosphorylation are mutated to alanine, the binding interaction between SMO and PKA-C is greatly diminished8.
This thesis describes experiments to better understand the structural and biochemical basis of the SMOct/PKA-C binding interaction.We hypothesize that GRK2/3 phosphorylation of SMOct will increase the protein’s binding affinity to PKA-C through ionic interactions created by the negatively charged phosphate. It is unclear whether phosphorylated SMOct binds directly to PKA-C, or if this phosphorylation promotes binding of an intermediary protein that bridges SMOct and PKA-C. To test these two models using an in vitro system, phosphomimetic mutants of SMOct were used in a competition binding assay with PKA-C and compared to wildtype SMOct to determine if phosphorylation decreases the Kd for the SMOct/PKA-C interaction. We utilized phosphomimetic mutation as opposed to GRK2/3 phosphorylation or phosphopeptide synthesis due to the poor ability of GRK2/3 to phosphorylate soluble substrates, and the relative difficulty of phosphopeptide synthesis in comparison to mutation. GRK2/3 phosphorylation of soluble substrates is difficult because GRK2/3 requires binding to the proximal side of the transmembrane domain of the active GPCR, along with membrane lipids (such as PIP2), which then allosterically activates GRK2/3 to phosphorylate the soluble GPCR domains. In the absence of the GPCR’s transmembrane domain, such as in SMOct, phosphorylation is highly inefficient in vitro and the turnover number (kcat/Km) decreases 1000-fold when compared with full length
GPCRs. We avoided using full-length SMO due to challenges associated with purifying and solubilizing the membrane protein for binding assays. The SMOct/PKA-C binding interaction is difficult to crystallize for structural analysis due to the weak binding affinity of the two proteins and largely unstructured nature of the SMOct. We tried to overcome this weak binding interaction by crosslinking the SMOct/PKA-C complex to add stability through a covalent bond. We optimized the crosslinking protocol and ran on a large scale to obtain a crosslinked complex that was sent to our collaborators for crystallography.
To obtain structural information about the SMOct peptide, we used NMR as this technique is the only high-resolution atomic-level approach that will allow information about unstructured proteins. NMR has been used in the past to obtain information about unstructured proteins similar to SMOct such as full-length PKI12. I optimized a protocol to express and purify isotopically labeled SMOct to be used in NMR analysis for structural information about the peptide.
Preliminary studies indicate that phosphomimetic mutations did not produce an effect on Kd, which may mean that phosphorylation does not increase binding affinity, though could also reflect technical limitations of the approach I am using. Crosslinking SMOct to PKA-C via a disulfide bond did not produce a crystallizable complex implying that other regions of the protein may need to be stabilized to facilitate crystal formation. These results will help inform ongoing experiments to better understand the SMOct/PKA-C binding interaction and capture this important complex for the first time.
METHODS
SMOct WT and phosphomimetic mutant purification:
SMOct WT and mutant peptides were purified in parallel using a bacterial expression system. An inducible strain of Escherichia coli, BL21(DE3), was transduced with the plasmid vector pHTSHP containing SMOct with N-terminal tags MBP-His8-SUMO. The vector pHTSHP also contains an ampicillin resistance gene to allow for selection of transduced cells. Following outgrowth for 1 hour at 37 degrees Celsius in SOC recovery media to allow for expression of ampicillin resistance, the cells were then grown overnight at 37 degrees Celsius in LB containing ampicillin to select for cells expressing the transduced plasmid. 5 mL of the overnight cultures were inoculated into 1 L of LB with ampicillin and grown at 37 deg Celsius until optical density, measured at 600 nm, of 1.0 was reached. Isopropyl β-D-1 thiogalactopyranoside (IPTG) applied to the culture induced the T7 promoter-based expression system to express the SMOct peptide. After induction, the temperature was decreased to 30 degrees Celsius and cells were grown for 5 hours. Cells were then pelleted using centrifugation at 5000 x g for 30 minutes and flash-frozen using liquid nitrogen.
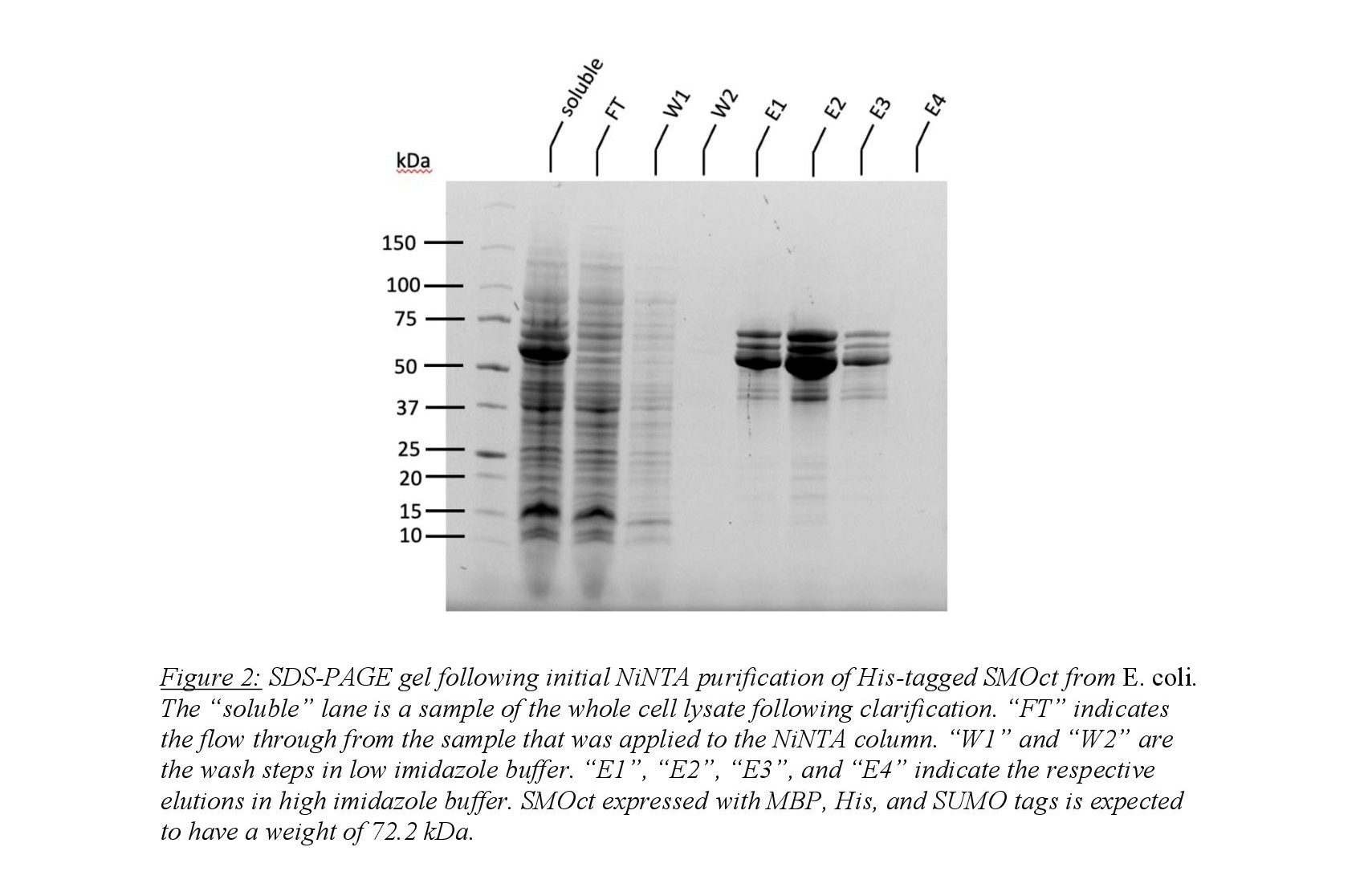
Purification of SMOct from the cell pellet began with resuspension in binding buffer (50mM Tris pH 8, 300 mM NaCl, 25 mM imidazole, 0.2 mM TCEP) followed by cell lysis via sonication in addition to lysozyme and benzonase. The whole cell lysate was clarified via centrifugation and the soluble fraction was isolated. Affinity chromatography using Ni-NTA resin was used to isolate the His8-tagged SMOct peptide. Wash steps used a buffer containing 25 mM imidazole to remove nonspecifically bound proteins on the Ni-NTA resin. Elution of tagged SMOct was performed using 250 mM imidazole and desired fractions were pooled (Figure 2). Imidazole was then dialyzed from pooled fractions using a two-step dialyzation process. MBP-His8-SUMO tags were cleaved using the protease ULP1 which cleaves the protein at the C-terminal domain of SUMO leaving SMOct untagged. Isolation of untagged SMOct was performed using a three-step process. First, the cleaved solution was applied to Ni-NTA column to isolate untagged SMOct using the buffers described above. The desired fractions were pooled and cation-exchange chromatography was performed to isolate SMOct from remaining impurities. Finally, gel filtration was used to isolate SMOct from SMOct degradation products.
PKA-C purification
Human PKA-C (hPKA-C) and mouse PKA-C (mPKA-C) orthologs were expressed using a bacterial expression system. BL21(DE3)-pLysS cells were transduced with a plasmid containing either hPKA-C (with N-terminal His6-SUMO tags) or mPKA-C (with no tags). The addition of the N-terminal tags improves expression of the protein as it is foreign to E. coli and is prone to proteolysis. Following outgrowth for 1 hour at 37 degrees Celsius in SOC recovery media to allow for expression of ampicillin and chloramphenicol resistance, the cells were grown overnight at 37 degrees Celsius in LB containing both antibiotics to select for cells expressing the transduced plasmid as well as the pLysS plasmid. 50 mL of the overnight culture were inoculated into 1 L of LB with both antibiotics and grown at 37 deg Celsius until optical density, measured at 600 nm, of 1.0 was reached. IPTG was used to induce the T7 promoter-based expression system to express the respective PKA-C ortholog. After induction, the temperature was decreased to 18 degrees Celsius and cells were grown overnight. Cells were then pelleted the following day using centrifugation at 5000 x g for 30 minutes and flash-frozen using liquid nitrogen.
The cell pellet was resuspended in TMN50 buffer (Tris 50 mM pH 7, 50 mM NaCl, 2 mM MgCl2, 0.4 mM ATP) followed by cell lysis via sonication. The lysed cell solution was
clarified using high-speed centrifugation (40,000 x g) for 30 minutes. The clarified sample was incubated with a shortened PKI peptide (IP20) bound to a resin for 90 minutes in binding conditions (400 mM ATP, 1 M MgCl2). IP20 binds to PKA-C with nanomolar affinity and is highly selective to binding PKA-C making it effective in isolating PKA-C from the clarified cell lysate. Incubated sample was applied to a column and washed with TMN50 and TMN250 (Tris 50 mM pH 7, 250 mM NaCl, 2 mM MgCl2, 0.4 mM ATP). PKA-C was then eluted from the IP20 resin using a high arginine buffer to displace the bound PKA-C from the column in 1 mL fractions using Bradford Reagent to monitor when the protein was completely eluted from the column. Fractions were run on an SDS-PAGE gel and PKA-C fractions were pooled.
hPKA-C contains His6-SUMO N-terminal tags that were then cleaved using UPL1 during dialysis to remove arginine from the buffer. mPKA-C is untagged and dialysis was performed in parallel without addition of a protease.
Isotopically labeled SMOct preparation:
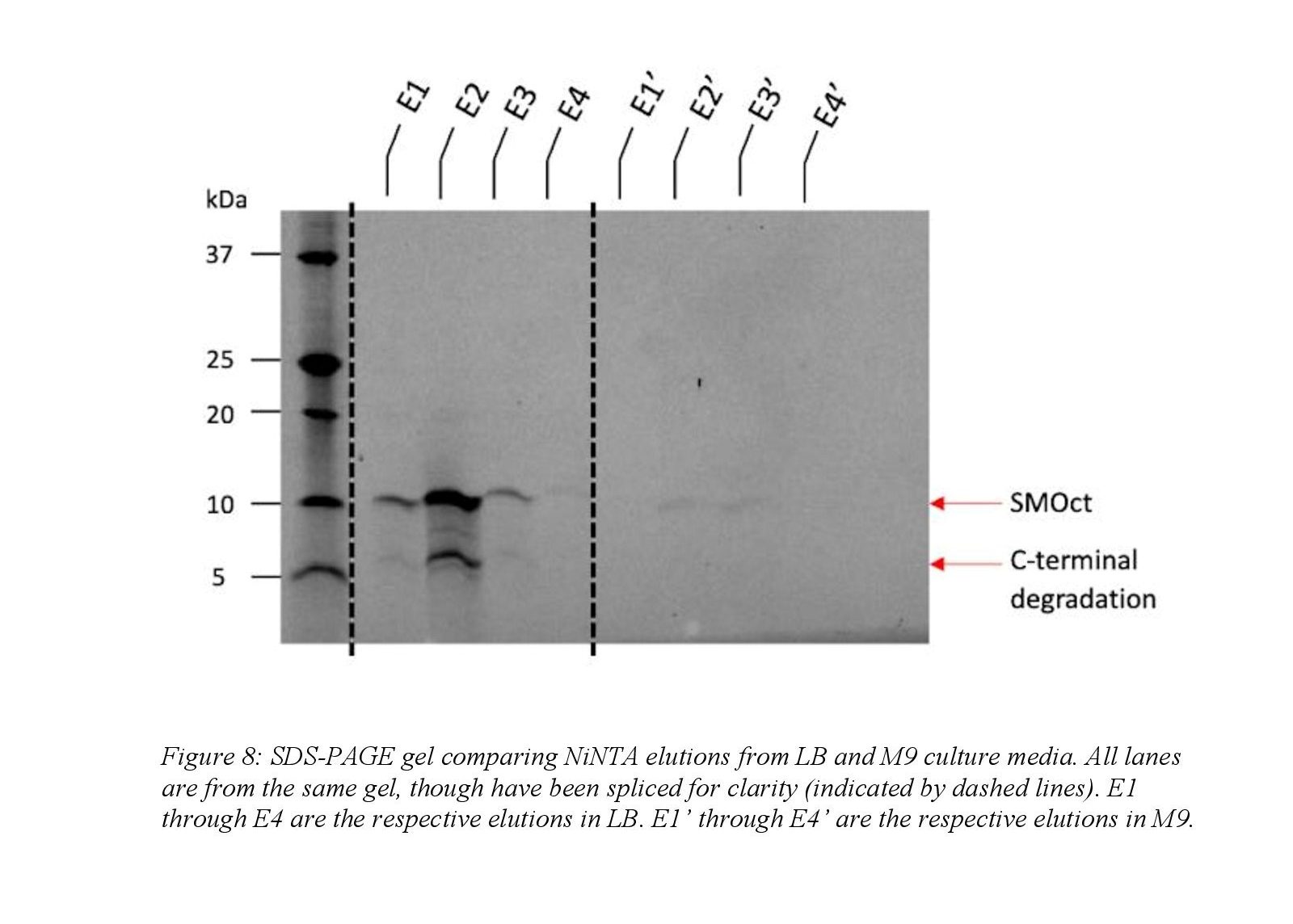
Isotopically SMOct was prepared similarly to SMOct grown in LB as described above and only notable differences will be outlined. The overnight growth following transduction was in LB, but minimal media (M9) was used during the outgrowth phase. Minimal media was necessary because it is chemically defined and isotopic labeling can be controlled by simply adding heavy isotopes which is not possible in rich media. The overnight LB culture was diluted into M9 media to an OD 600 of 0.1 and induced with IPTG at OD 600 1.0. Following clarification of the lysed cell pellet, the soluble fraction obtained was boiled to denature the soluble, structured proteins and clarified at 40,000 x g to remove the precipitated proteins. The soluble fraction remaining was purified using Ni-NTA affinity chromatography as described above. Ion exchange and gel filtration was not performed on the sample after Ni-NTA chromatography.
Fluorescence Polarization Assay:
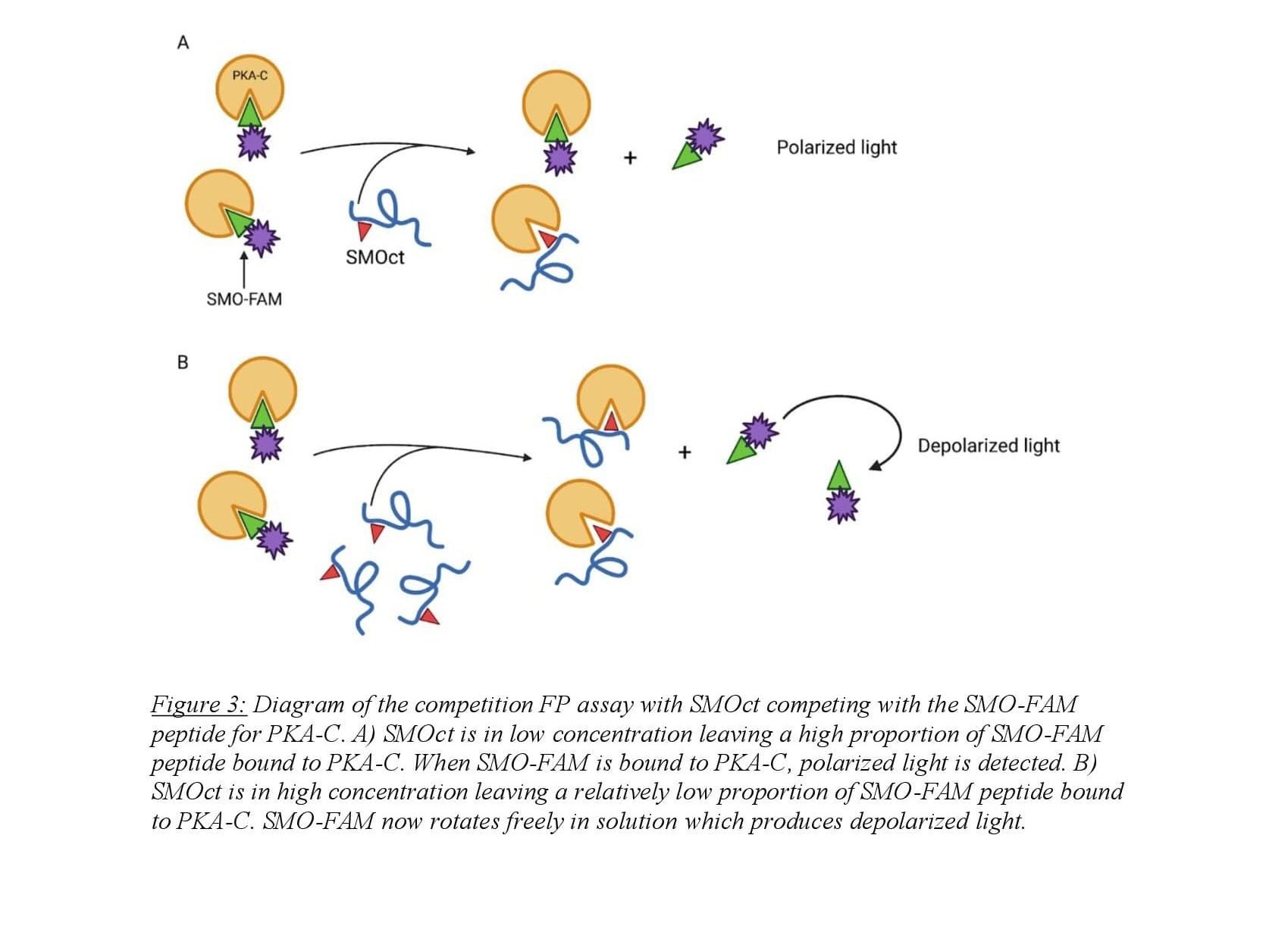
Fluorescence polarization (FP) is a biochemical technique used to quantitatively determine molecular binding interactions. The assay utilizes a fluorophore-bound molecule competing with an untagged molecule for a common binding partner. Polarized light is applied to the sample and excites the fluorophore which emits polarized light proportional to the amount of fluorophore bound to its binding partner which tumbles in solution slowly. When the fluorophore is competed from the binding partner, it rotates in solution rapidly emitting depolarized light when excited by light applied to the sample13. In our case, protein-protein interactions are monitored using a Fluorescin (FAM) tagged, truncated SMOct peptide in competition with untagged SMOct peptide binding with PKA-C (Figure 3). FAM-tagged SMOct and PKA-C were kept at constant concentrations of 40 nM and 3.5 μM respectively. This concentration of FAM-tagged SMOct was chosen to adequately detect changes in polarization while also being low enough for SMOct to compete with the peptide for the PKA-C binding site. The untagged SMOct peptide was serially diluted creating ten SMOct samples with a high concentration of 24 μM to maximally bind the available PKA-C.
The reaction was monitored by measuring polarization using a Tecan plate reader with readings at 0 min, 5 min, 15 min, and 30 min. Multiple readings were taken at the indicated time points to ensure that the reactions were at equilibrium and to monitor any variation between readings. Each experiment was carried out in at least duplicate with FAM readings at an excitation wavelength of 485 nm and emission wavelength of 535 nm. Using the absorbance data, a dose-response curve was created and IC50 was calculated. This experiment allowed us to compare the binding affinity of SMOct/PKA-C with that of phosphomimetic SMOct/PKA-C.
Crosslinking:
Crosslinking the SMOct/PKA-C complex was employed for structural analysis of the protein-protein binding interaction. SMOct was mutated to contain a cysteine residue at amino acid 637 which is at the P+2 site. Mutation at the P+2 site was chosen for two reasons. First, when this residue was mutated to alanine, binding still occurred between SMOct and PKA-C indicating that mutation to cysteine will not eradicate the binding interaction9. Second, Susan Taylor’s lab has mutated the P+2 site on the protein kinase regulatory subunit II to cysteine and crosslinked with a cysteine present in the adjacent binding groove of PKA-C for use in structural studies14. Human PKA-C contains two cysteine residues so to prevent unwanted intramolecular crosslinking, the cysteine at amino acid 343 was mutated to a serine. Purification of the SMOct-(L637C) was carried out as described above for SMOct-WT. Purification of hPKA-C-(C343S) was performed as described above from hPKA-C. Researchers Annabel Lee and Corvin Arveseth optimized the crosslinking on a small scale prior to performing the reaction on a large scale. This included varying the concentration of the two mutant proteins, time, temperature, and pH until efficient crosslinking was observed.
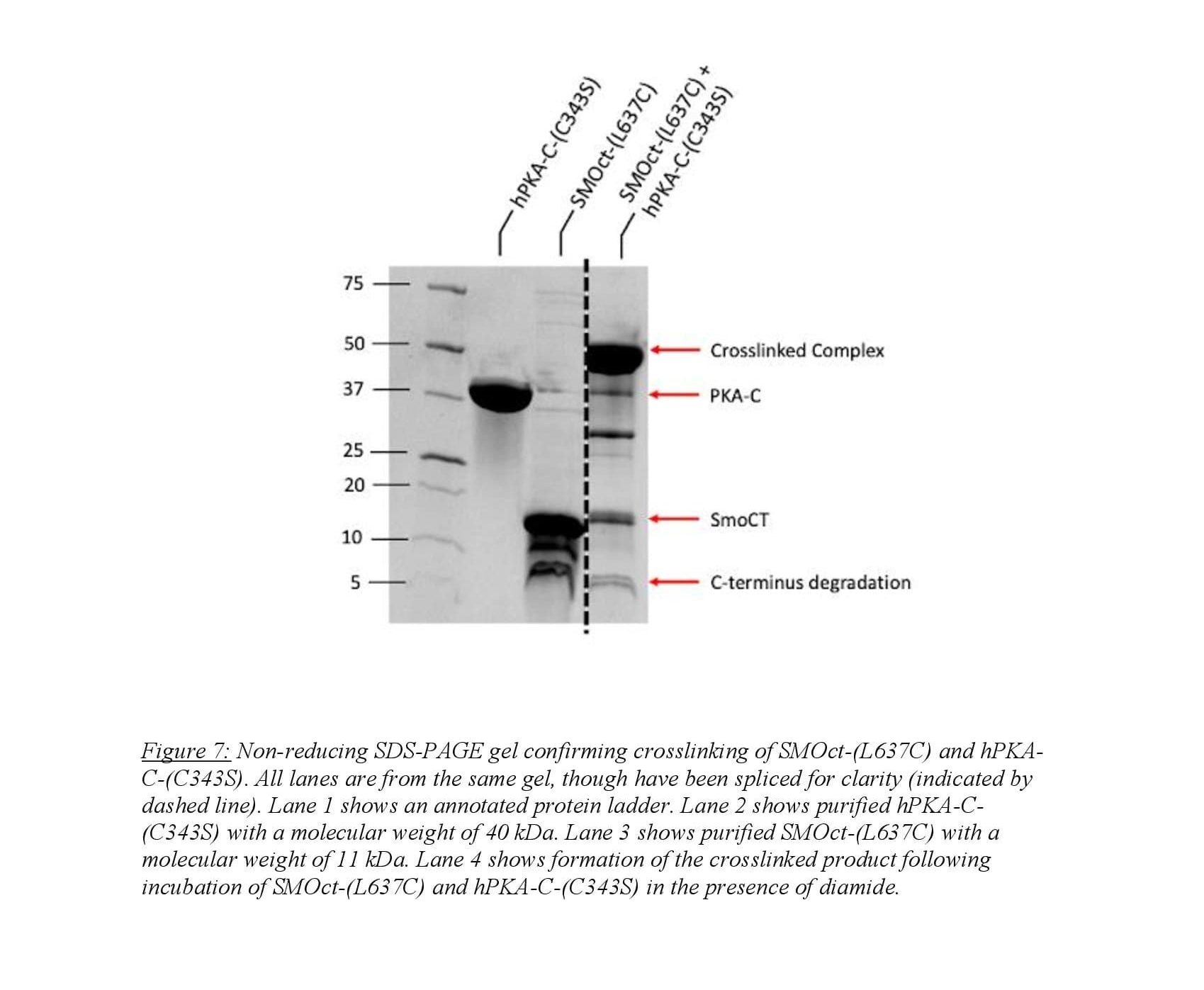
Large-scale crosslinking involved buffer exchange of SMOct-(L637C) and hPKA-C-(C343S) into a non-oxidizing buffer. Samples were then combined at desired concentrations and incubated for 30 minutes with ATP and MgCl2 to induce SMOct/PKA-C complex. The oxidizing agent diamide was then applied to the sample and incubated for 3 hours to induce cysteine-cysteine crosslink. The product was then run on a non-reducing SDS-PAGE gel to confirm crosslinked product formation.
RESULTS
Phosphomimetic SMOct competition FP assay:
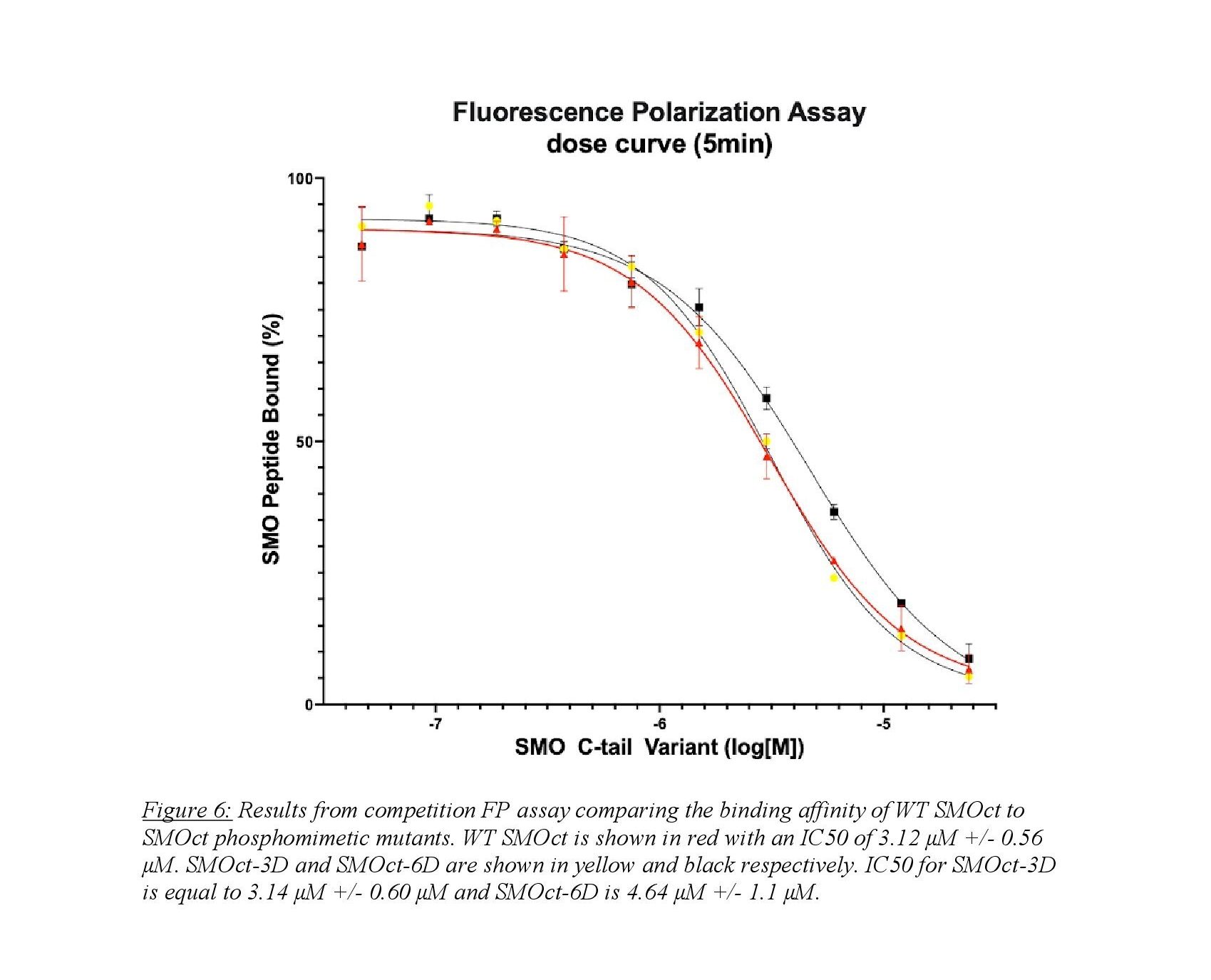
A competition FP assay is able to provide quantitative data in an in vitro system. Figure 6 shows the results from a competition FP assay between SMOct-WT and two phosphomimetic SMOct mutants. The SMOct-3D contains mutations to aspartic acid (D) at known GRK2/3 phosphorylation sites within the B cluster (Figure 5). The SMOct-6D mutant contains the same three mutations in the B cluster, but also includes 3 additional aspartic acid mutations at the C cluster.
We hypothesized that these mutations would mimic the effect of a negative charge similar to a phosphate. It is unclear whether the phosphorylated SMOct/PKA-C complex relies on the ionic interactions from having negatively charged phosphates, or if the addition of phosphates provides another role in the protein binding interaction such as steric bulk or an alternate conformation of SMOct. The FP assay provides quantitative data and allows for comparison of binding affinity via the half-maximal inhibitory concentration (IC50). This is a measure of potency and describes what concentration of SMOct is necessary to displace half the fluorescent peptide from the active site of PKA-C. As seen in Figure 6, we do not see a significant difference in IC50 values between the SMOct-3D mutant and SMOct-WT. This suggests that the presence of negative charges at the B cluster is not sufficient for increased binding affinity between SMOct and PKA-C. It is also possible that an aspartic acid residue does not adequately mimic the size and conformation of a phosphoryl group. The tetrahedral geometry of the phosphoryl group might be necessary in the binding interaction, which would not be well represented by a carboxylate group on an aspartate.
SMOct/PKA-C crosslinking:
Structural analysis of the SMOct/PKA-C complex is difficult to perform due to the low binding affinity between the two proteins, which hinders stable complex formation9. Because of this complication, our understanding of the binding interaction has relied on analogies to existing complexes between PKA-C and high-affinity pseudosubstrates, which are quite different from SMO as explained earlier. To directly characterize the SMO/PKA-C complex in structural terms, we must first overcome the low binding affinity. To this end we employed cysteine crosslinking to covalently link the protein complex for crystallography studies for comparison to predicted models.
Our work focused on the purification and crosslinking of SMOct-(L637C) and hPKA-C-(C343S). As discussed in “Methods,” we crosslinked the SMOct mutant to hPKA-C using diamide as the oxidizing agent. Annabel Lee and Isaac Nelson of the Myer’s lab optimized the protocol for crosslinking using a small scale binding assay testing various conditions and concentrations of SMOct and PKA-C. We used the optimized protocol on a larger scale to obtain a sufficient sample for crystallography studies. We confirmed the results of the crosslinking using gel electrophoresis (Figure 7). This shows a clear band at 51 kDa that corresponds to the summation of hPKA-C-(C343S) and SMOct-(L637C) which weigh 40 kDa and 11 kDa respectively. We used gel filtration to further purify the crosslinked complex from remaining protein and degradation products. The crosslinked complex was sent to Jessica Bruystens at UCSD for crystallography studies. Several thousand different conditions were tested to crystallize the crosslinked complex and none were successful indicating that there is essentially no way that this complex will crystallize without further increasing the stability.
Isotopically labeled SMOct:
Preparations to optimize the protocol for purification of the isotopically labeled SMOct proved that the expression in minimal media is challenging. Because SMOct is foreign to E. coli, the protein is toxic to the cells and largely inhibits cell growth in minimal media. SMOct expression improved when BL21(DE3) pLysS E. coli cells were used to minimize basal expression of SMOct prior to IPTG induction. Even with decreasing basal expression of SMOct, cell growth in M9 media to reach an OD 600 of 1.0 was variable and took 10 to 15 hours which differed greatly from growth in LB where the culture reached the same density in approximately 3 to 5 hours. The yield of SMOct expressed in M9 also decreased compared to LB by a factor of 6. Decreased expression between the LB and M9 media is seen qualitatively in the elution fractions during NiNTA (Figure 8). Thus, preliminary studies demonstrate that expression in the traditional M9 medium is too low to support NMR analysis, and further optimization is needed to purify isotopically labeled SMOct.
DISCUSSION
Phosphomimetic SMOct competition FP assay:
The competition FP binding assay was used to test the hypothesis that phosphorylated SMOct would bind with higher affinity to PKA-C than SMOct without phosphorylation. Because phosphorylation of the cytoplasmic tail via GRK2/3 requires the seven transmembrane domain of SMO, we were unable to use direct phosphorylation of SMOct and rather mutated known GRK2/3 phosphorylation sites to aspartic acid. Our results indicate that there is no significant change in binding affinity between wildtype SMOct and SMOct-3D, with decreased binding affinity of the SMOct-6D mutant, though the decrease in IC50 from WT is not statistically significant. These results contradict our initial hypothesis and predicted models of the SMOct/PKA-C complex. However, there are possible explanations for this finding. First, the size and shape of the phosphoryl group at the GRK2/3 phosphorylation site could have a role in promoting the correct conformation for SMOct to bind PKA-C. Because the carboxyl group of the aspartic acid side chain is relatively smaller and with a different geometry than a phosphoryl group, it might not mimic the effects of phosphorylation completely. Second, the charge of the phosphoryl group is -1.5 whereas the charge of an aspartate residue is -1.0 at physiologic pH15. This difference in charge between the aspartate residue and phosphoryl group could explain the lack of increased affinity given the importance that the ionic interaction plays in the predicted model. Based on these caveats, the experiments with the phosphomimetic mutations are inconclusive, because we cannot be sure that these mutations accurately reflect the effects of GRK2/3 phosphorylation on the SMOct/PKA-C binding interaction. In addition, mass spectrometry of purified SMO from mammalian cells shows that phosphoryl groups do not necessarily occupy all phosphorylation sites on SMOct at the same time8. It is possible that having three, or six, phosphomimetic mutations on SMOct actually destabilize the conformation required to bind PKA-C. To test this hypothesis in future studies, single phosphomimetic mutations at the known GRK2/3 phosphorylation sites will be used in the same binding assay to determine if this affects binding with PKA-C.
Additional experimentation to overcome the difficulty with GRK2/3 phosphorylation and complications with phosphomimetics will include synthesis of the SMOct with phosphoserine or phosphothreonine at the GRK2/3 phosphorylation sites. This would effectively overcome all the issues presented by in vitro GRK2/3 phosphorylation and phosphomimetic mutation of the SMOct. Because the SMOct is 110 amino acids in length, synthesis of the peptide using solid phase peptide synthesis poses its own set of challenges which is why we did not pursue this route prior to phosphomimetics. However, given the inconclusive nature of our phosphomimetic mutant analysis, we will now attempt to synthesize the phosphopeptide in collaboration with Michael Kay, whose lab has extensive expertise with synthetic peptide synthesis. The synthetic phosphopeptide could then be used in a binding assay with PKA-C and compared to WT SMOct to determine the effects of phosphorylation on SMOct binding affinity.
Another possible experiment to overcome the difficulties presented is to phosphorylate full length SMO in detergent micelles directly with GRK2/3 in the presence of the membrane lipid PIP2 (which allosterically activates GRK2/3, see Introduction). This approach allows GRK2/3 to dock to the intracellular domain of SMO and the membrane lipid PIP2 to then phosphorylate SMOct which is not possible with soluble substrates such as SMOct. Other members of the Myers lab are currently working on a protocol for in vitro phosphorylation of SMO which could then potentially be used in a similar binding assay to determine the effects of direct phosphorylation in binding with PKA-C.
SMOct/PKA-C structural analysis:
Structural analysis of the SMOct/PKA-C complex proves to be difficult via conventional structural biology methods. This is due to the transient nature of the binding interaction which makes techniques such as crystallography and cryogenic electron microscopy difficult. We hypothesized that covalently crosslinking the complex via a disulfide bond would stabilize the two proteins and enable crystallization. My work focused on the preparation of the protein complex which was sent to Susan Taylor’s lab at UCSD for crystallography. Despite modification to covalently bond the two proteins, the SMOct/PKA-C complex did not crystallize and we were unable to obtain structural information from this experiment. This result suggests that the protein complex requires further stabilization to crystallize. Possible ways to stabilize the complex include additional covalent bond attachments, truncation of the SMOct peptide to remove unstable regions, or addition of antibodies/nanobodies that support the bound complex.
Another route I am exploring to obtain structural information regarding SMOct is through NMR due to the ability to obtain information about unstructured proteins at an atomic level. Structural information about the binding interaction of SMOct/PKA-C can be observed through chemical shift perturbations in the NMR amide fingerprint of isotopically labeled SMOct. This technique was used to pinpoint the residues on PKA-C that are involved in binding to SMOct, though it has not been performed with isotopically labeled SMOct.
My work is currently focused on optimizing a protocol to express isotopically labeled SMOct in E. coli. Minimal media is required to grow the cells to ensure the culture only contains isotopically labeled nitrogen (15N). Expression levels of SMOct in minimal media continue to remain low despite attempts to decrease basal protein expression prior to induction. This is likely due to a combination of factors including SMOct toxicity to E. coli as well as decreased rate of bacterial growth in minimal media. One possible method to overcome this difficulty in expression is through autoinduction. This method of bacterial protein expression utilizes a culture medium containing both glucose and lactose. Glucose is preferentially used as an energy source by E. coli, but once the glucose in the media is consumed, lactose is then metabolized producing allolactose and activating expression of target genes via the lac operon. This protocol offers advantages over IPTG induction for toxic proteins as the bacteria are able to self-regulate protein expression levels. This allows for gradual protein expression that can prevent the deleterious effects of toxic proteins on bacterial growth16. This method is encouraging for growth of isotopically labeled SMOct given its success in the past with SMOct truncated peptides and with other recombinant proteins that are toxic to bacteria.
Ciliary regulation of SMO:
To explain why the SMOct/PKA-C binding interaction is a low affinity interaction and relevant in PKA-C sequestration by SMO, physiologic conditions have to be considered. The primary cilium is essential for Hh signal transduction17. This is due to the compartmentalization of SMO which, when activated, accumulates 20-fold in the primary cilium18. This increase in the local concentration of SMO allows for regulation of a smaller pool of ciliary PKA-C, as most cytoplasmic PKA-C is inactive and bound in its holoenzyme form with protein kinase A regulatory subunits (PKA-R). This is an important regulatory mechanism of the Hh pathway as PKA is used in myriad cellular processes19. SMOct does not bind to the PKA holoenzyme8, as binding is specific to the PKA-C active site which is not available when PKA-R is bound. This compartmental regulation of PKA-C is physiologically important as it allows Hh pathway activation to target GLI without disrupting PKA-dependent processes throughout the cell20. It also offers insight into how the SMO/PKA-C interaction, while of only modest affinity, may nevertheless be highly relevant during Hh signal transduction in its native ciliary environment.
Because SMO is acting on a smaller, defined pool of ciliary PKA-C, small changes in free PKA-C concentration have the potential to create large effects given that gene expression is the final target of the Hh pathway. Continuing to improve understanding of this low affinity interaction between SMOct and PKA-C through biochemical and structural studies will be vital to understanding the complete mechanistic details of Hh signal transduction.


REFERENCES
(1) Nüsslein-Volhard, C.; Wieschaus, E. Mutations Affecting Segment Number and Polarity in Drosophila. Nature 1980, 287 (5785), 795–801. https://doi.org/10.1038/287795a0.
(2) Pak, E.; Segal, R. A. Hedgehog Signal Transduction: Key Players, Oncogenic Drivers, and Cancer Therapy. Dev. Cell 2016, 38 (4), 333–344. https://doi.org/10.1016/j.devcel.2016.07.026.
(3) Taipale, J.; Cooper, M. K.; Maiti, T.; Beachy, P. A. Patched Acts Catalytically to Suppress the Activity of Smoothened. Nature 2002, 418 (6900), 892–897. https://doi.org/10.1038/nature00989.
(4) Zhang, Y.; Bulkley, D. P.; Xin, Y.; Roberts, K. J.; Asarnow, D. E.; Sharma, A.; Myers, B. R.; Cho, W.; Cheng, Y.; Beachy, P. A. Structural Basis for Cholesterol Transport-like Activity of the Hedgehog Receptor Patched. Cell 2018, 175 (5), 1352-1364.e14. https://doi.org/10.1016/j.cell.2018.10.026.
(5) Deshpande, I.; Liang, J.; Hedeen, D.; Roberts, K. J.; Zhang, Y.; Ha, B.; Latorraca, N. R.; Faust, B.; Dror, R. O.; Beachy, P. A.; Myers, B. R.; Manglik, A. Smoothened Stimulation by Membrane Sterols Drives Hedgehog Pathway Activity. Nature 2019, 571 (7764), 284–288. https://doi.org/10.1038/s41586-019-1355-4.
(6) Niewiadomski, P.; Kong, J. H.; Ahrends, R.; Ma, Y.; Humke, E. W.; Khan, S.; Teruel, M. N.; Novitch, B. G.; Rohatgi, R. Gli Protein Activity Is Controlled by Multisite Phosphorylation in Vertebrate Hedgehog Signaling. Cell Rep. 2014, 6 (1), 168–181. https://doi.org/10.1016/j.celrep.2013.12.003.
(7) Low, W.-C.; Wang, C.; Pan, Y.; Huang, X.-Y.; Chen, J. K.; Wang, B. The Decoupling of Smoothened from Galphai Proteins Has Little Effect on Gli3 Protein Processing and Hedgehog-Regulated Chick Neural Tube Patterning. Dev. Biol. 2008, 321 (1), 188–196. https://doi.org/10.1016/j.ydbio.2008.06.014.
(8) Arveseth, C. D.; Happ, J. T.; Hedeen, D. S.; Zhu, J.-F.; Capener, J. L.; Shaw, D. K.; Deshpande, I.; Liang, J.; Xu, J.; Stubben, S. L.; Nelson, I. B.; Walker, M. F.; Kawakami, K.; Inoue, A.; Krogan, N. J.; Grunwald, D. J.; Hüttenhain, R.; Manglik, A.; Myers, B. R. Smoothened Transduces Hedgehog Signals via Activity-Dependent Sequestration of PKA Catalytic Subunits. PLOS Biol. 2021, 19 (4), e3001191. https://doi.org/10.1371/journal.pbio.3001191.
(9) Happ, J. T.; Arveseth, C. D.; Bruystens, J.; Bertinetti, D.; Nelson, I. B.; Olivieri, C.; Zhang, J.; Hedeen, D. S.; Zhu, J.-F.; Capener, J. L.; Bröckel, J. W.; Vu, L.; King, C. C.; Ruiz-Perez, V. L.; Ge, X.; Veglia, G.; Herberg, F. W.; Taylor, S. S.; Myers, B. R. A PKA Inhibitor Motif within SMOOTHENED Controls Hedgehog Signal Transduction. Nat. Struct. Mol. Biol. 2022, 29 (10), 990–999. https://doi.org/10.1038/s41594-022-00838-z.
(10) Liu, C.; Ke, P.; Zhang, J.; Zhang, X.; Chen, X. Protein Kinase Inhibitor Peptide as a Tool to Specifically Inhibit Protein Kinase A. Front. Physiol. 2020, 11, 574030. https://doi.org/10.3389/fphys.2020.574030.
(11) Knape, M. J.; Ballez, M.; Burghardt, N. C.; Zimmermann, B.; Bertinetti, D.; Kornev, A. P.; Herberg, F. W. Divalent Metal Ions Control Activity and Inhibition of Protein Kinases. Metallomics 2017, 9 (11), 1576–1584. https://doi.org/10.1039/c7mt00204a.
(12) Olivieri, C.; Wang, Y.; Li, G. C.; V S, M.; Kim, J.; Stultz, B. R.; Neibergall, M.; Porcelli, F.; Muretta, J. M.; Thomas, D. D.; Gao, J.; Blumenthal, D. K.; Taylor, S. S.; Veglia, G. Multi-State Recognition Pathway of the Intrinsically Disordered Protein Kinase Inhibitor by Protein Kinase A. eLife 2020, 9, e55607. https://doi.org/10.7554/eLife.55607.
(13) Moerke, N. J. Fluorescence Polarization (FP) Assays for Monitoring Peptide-Protein or Nucleic Acid-Protein Binding. Curr. Protoc. Chem. Biol. 2009, 1 (1), 1–15. https://doi.org/10.1002/9780470559277.ch090102.
(14) First, E. A.; Taylor, S. S. Induced Interchain Disulfide Bonding in CAMP-Dependent Protein Kinase II. J. Biol. Chem. 1984, 259 (7), 4011–4014.
(15) Dephoure, N.; Gould, K. L.; Gygi, S. P.; Kellogg, D. R. Mapping and Analysis of Phosphorylation Sites: A Quick Guide for Cell Biologists. Mol. Biol. Cell 2013, 24 (5), 535–542. https://doi.org/10.1091/mbc.E12-09-0677.
(16) Blommel, P. G.; Becker, K. J.; Duvnjak, P.; Fox, B. G. Enhanced Bacterial Protein Expression During Auto-Induction Obtained by Alteration of Lac Repressor Dosage and Medium Composition. Biotechnol. Prog. 2007, 23 (3), 585–598. https://doi.org/10.1021/bp070011x.
(17) Gigante, E. D.; Caspary, T. Signaling in the Primary Cilium through the Lens of the Hedgehog Pathway. Wiley Interdiscip. Rev. Dev. Biol. 2020, 9 (6), e377. https://doi.org/10.1002/wdev.377.
(18) Corbit, K. C.; Aanstad, P.; Singla, V.; Norman, A. R.; Stainier, D. Y. R.; Reiter, J. F. Vertebrate Smoothened Functions at the Primary Cilium. Nature 2005, 437 (7061), 1018–1021. https://doi.org/10.1038/nature04117.
(19) Bangs, F.; Anderson, K. V. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb. Perspect. Biol. 2017, 9 (5), a028175. https://doi.org/10.1101/cshperspect.a028175.
(20) Tuson, M.; He, M.; Anderson, K. V. Protein Kinase A Acts at the Basal Body of the Primary Cilium to Prevent Gli2 Activation and Ventralization of the Mouse Neural Tube. Development 2011, 138 (22), 4921–4930. https://doi.org/10.1242/dev.070805.
