Science
103 Investigating the Role of C5orf42 in Diabetic Kidney Disease
Karah Hall
Faculty Mentor: Marcus Pezzolesi (School of Biological Sciences, University of Utah)
ABSTRACT
Diabetic kidney disease is a complication of diabetes and is the world’s leading cause of end- stage kidney disease. Genetic factors are known to contribute to diabetic kidney disease susceptibility, however, despite intense effort, the identification of variants that underlie its risk has been challenging. Through the analysis of four pathogenic variants, we were able to investigate the potential role of the ciliary gene C5orf42 in the susceptibility of kidney disease in patients with diabetes. C5orf42 variants (I165Yfs*17 and S123F) were found to localize to different alleles, meaning that the biallelic carrier was a compound heterozygous carrier. C5orf42 variant V938Efs*27 was found to co-segregate with disease in a nuclear family. Lastly, C5orf42 variant c.8300-G>C was found to be associated with three different isoforms.
INTRODUCTION
Diabetic kidney disease (DKD), also diabetic nephropathy, is a complication of diabetes that damages the blood vessels or glomeruli of the kidney and, therefore, affects the kidney’s ability to remove waste products from the body. DKD is also the world’s leading cause of end-stage kidney disease, which is life-threatening and has limited treatment options that include dialysis and/or kidney transplantation. Identification of the genetic factors that contribute to DKD’s risk may help identify those at risk of this disease and potentially lead to improved treatment or prevention strategies; these processes have been challenging thus far.11
The first published genome-wide association study on DKD susceptibility, published in 2009, was the Genetics of Kidneys in Diabetes (GoKinD) study.3 Since then, there have only been a few reported genetic variants that have achieved genome-wide significance between single-nucleotide polymorphisms (SNPS) and this phenotype. Even fewer genetic variants have been replicated across different studies.1-6 Disappointingly, none of these findings have been able to help improve diagnosis or remediation tactics for patients at risk of DKD. However, there is evidence of its strong genetic predisposition, including an estimated heritability as high as 59%.5 Therefore, additional research is essential to better understand the genetic underpinnings of DKD and new approaches are necessary to increase the amount of gene discovery and make progress in this area of research.
With this goal in mind, Dr. Marcus Pezzolesi and his lab have used the Utah Diabetes Database, which contains electronic medical record data for more than 350,000 diabetic patients, the Utah Population Database, a unique population-based genealogy resource containing family histories and demographic data for 14 million individuals, and the Utah Kidney Study Biorepository, a biorepository of more than 2,000 diabetic and non-diabetic patients with kidney disease, to recently identify significant enrichment of 4 rare variants in C5orf42 that are predicted to be pathogenic, including 3 putative loss of function (pLOFs) variants, in patients with diabetes and end-stage kidney disease. They were able to do so through targeted next-generation sequencing using a custom panel comprising 345 kidney disease-related genes in 222 participants of the Utah Kidney Study, including 98 individuals with non-diabetic kidney disease and 124 individuals with DKD.7
Interestingly, 25% of DKD patients positive for pathogenic or likely pathogenic variants were found to carry rare variants with minor allele frequencies of less than 0.01 in known ciliopathy-associated genes, including 3 patients with type 2 diabetes and end-stage kidney disease found to harbor rare pLOF variants in C5orf42 (c.8300-1G>C, I165Yfs*17, and V938Efs*27). Among these patients, two C5orf42 variants (I165Yfs*17, a pLOF variant, and S123F, a variant predicted to be damaging by five of six computational prediction methods) were observed in one individual, suggesting that this patient could be a compound heterozygous carrier of rare damaging variants in this gene. Another carrier of a C5orf42 pLOF variant has a family history of diabetes and DKD and has had their family members recruited to the Utah Kidney Study to investigate whether these variants co-segregate with disease in this family.
C5orf42, also known as CPLANE1 and JBTS17, is associated with rare autosomal recessive ciliopathies characterized by cystic kidney disease.8-10 Importantly, in the presence of hyperglycemia, a state common in patients with diabetes, defects in cilia structure or function cause alterations in the kidney, including podocyte effacement, interstitial inflammation, and proteinuria.12 Based on these data, it can be hypothesized that defects in CPLANE1, the protein encoded by C5orf42, could contribute to DKD.
Although defects in ciliary genes have largely been associated with rare disease pathology, a recent study by Drivas and colleagues demonstrated the utility of a Mendelian pathway-based approach to genomic association studies linking variants in ciliopathy genes to common complex disease, including several kidney-related traits.13 This paradigm
shift represents a major milestone in our understanding of the role of the primary cilium, a sensory organelle found in nearly every human cell, in human disease and, importantly, supports the role of ciliary genes in kidney disease pathogenesis.
These novel discoveries suggest that C5orf42, which is associated with rare autosomal recessive ciliopathies characterized by cystic kidney disease8-10, may also contribute to DKD. Here, we expand on this finding by further investigating the potential role of four pathogenic variants in C5orf42 on the susceptibility of kidney disease in patients with diabetes.
METHODS
Sanger Sequencing-based Validation of C5orf42 Variants:
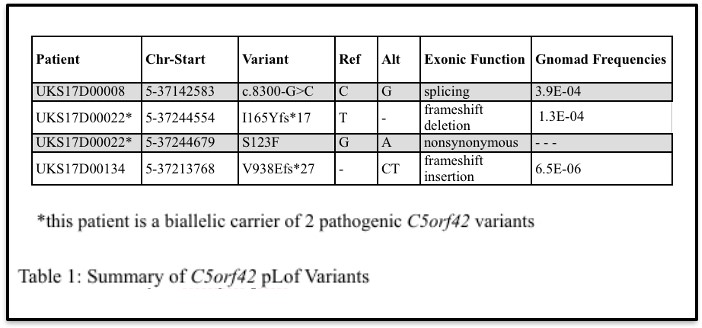
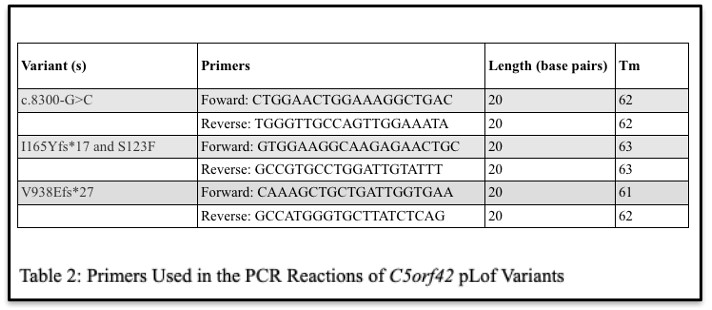
Four putative pathogenic variants in C5orf42 that were identified in 3 DKD patients using targeted next-generation sequencing were analyzed as part of this study (Table 1).
First, genomic DNA was isolated from whole blood from all C5orf42 variant carriers as well as recruited family members of UKS17D00134 using a standard phenol:chloroform DNA extraction protocol. Next, to confirm the presence of each variant in the carriers identified using next-generation sequencing, we preformed Sanger sequencing-based validation. PCR reactions were optimized to amplify each C5orf42 variant using the primers listed in Table 2. PCR amplicons were purified using ExoSAP-IT Express (Applied Biosystems, Waltham, MA) and sequenced using an Applied Biosystems 3730xl by the University of Utah’s DNA Sequencing Core. The resulting chromatograms were then analyzed using the Sequencer 5.4.6 software by Genes Codon Corporation.
Sub-Cloning of I165Yfs*17 and S123F Variants:
PCR amplicons from the carrier of biallelic C5orf42 variants (I165Yfs*17 and S123F) were also sub-cloned into E. coli using the TOPO TA cloning protocol with chemically competent TOP10 cells (Invitrogen, Waltham, MA) and plated on Luria Broth (LB) agar plates with ampicillin. Colonies were selected and inoculated in LB ampicillin media. Following inoculation, DNA was isolated using the QIAprep Miniprep Kit (Qiagen, Hilden, Germany) and Sanger sequenced to assess whether these variants localized to a single allele or to both alleles and whether this patient was a compound heterozygous carrier of these variants.
Segregation Analysis of the V938Efs*27 Variant:
In order to analyze the nuclear family of patient UKS17D00134 carrying the V938Efs*27 variant of C5orf42, isolated DNA was PCR amplified, purified, and submitted for Sanger sequencing. The resulting chromatograms were analyzed and segregation of this variant with diabetes and kidney disease were assessed in this family.
RNA Sequencing (RNASeq) Analysis:
To provide information about the transcriptome of each patient, total RNA was isolated from whole blood of carriers of each of the C5orf42 variants, including the family of the carrier of the V938Efs*27 variant using the PAXgene Blood RNA Kit (Qiagen, Hilden, Germany). A total of 7 samples were submitted to the University of Utah’s High-Throughput Sequencing Core for library preparation using NEBNext Ultra II Directional RNA Library Prep with rRNA Depletion Kit (New England Biolabs, Ipswich, MA) and RNA sequencing. The resulting data were analyzed using the Integrative Genomics Viewer (IGV) software.
Characterization of the c.8300-G>C Variant:
In order to authenticate observations made with IGV, which suggest multiple isoforms, PCR amplicons of the carrier of the splice variant c.8300-G>C were sub-cloned into E. coli, purified, and Sanger sequenced as described above. This variant was also further analyzed using the 3’ RACE System for Rapid Amplification of cDNA Ends (Invitrogen, Waltham, MA), which utilizes the natural poly(A) tail found in mRNA as a nonspecific priming site for PCR to characterize mRNA transcripts. After amplifying cDNA from patient UKS17D00008 using the 3’ RACE kit, the products were analyzed on an agarose gel and were also sub-cloned into E. coli.
These colonies were then Sanger sequenced.
The c.8300-G>C variant was further analyzed using the Genomis HSF Pro system, an online splice site prediction software. The genomic position of the variant was entered into the Mutation Analysis tool, which can detect a variant’s impact on splicing signals and acceptor and donor sites.
RESULTS
Confirmation of C5orf42 Variants in DKD Patients:
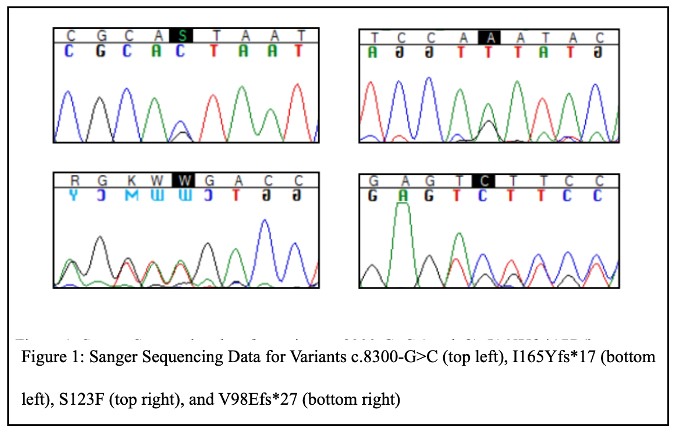
Our initial Sanger sequencing experiments confirmed our targeted next-generation sequencing results and that each DKD patient carried the rare C5orf42 variant observed in these data (Figure 1).
Evidence for Compound Heterozygosity:
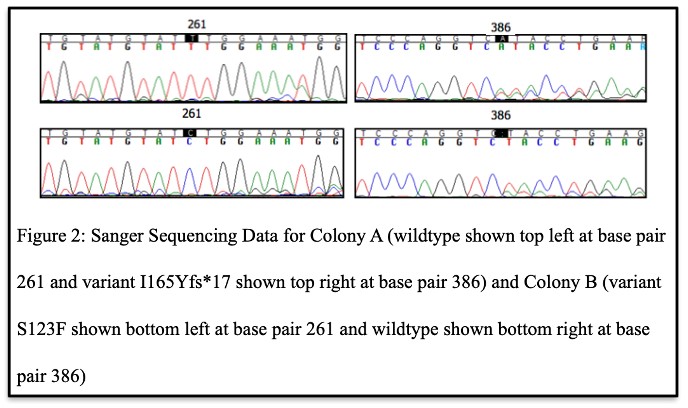
After the presence of the variants in each DKD patient was confirmed, the sequencing results of UKS17D00022, the carrier of biallelic C5orf42 variants (I165Yfs*17 and S123F), from the TOPO TA cloning procedure determined that some E. coli colonies displayed variant I165Yfs*17, and others displayed variant S123F (Figure 2). No colonies displayed both variants or no variation at all. Therefore, these data show that these variants localize to different alleles or different chromosomes, meaning that this individual is a compound heterozygous carrier of rare variants in C5orf42.
Co-segregation of Variant V98Efs*27 and DKD in Pedigree of UKS17D00134:
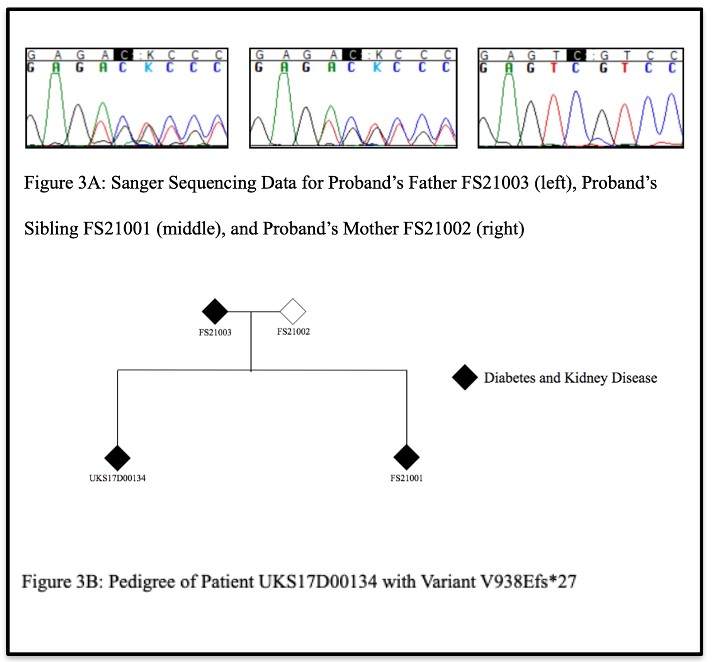
Next, Sanger sequencing results from the nuclear family of patient UKS17D00134, carrying the V938Efs*27 variant of C5orf42, revealed that in addition to the proband being a carrier of this variant, the proband’s father and sibling, both of whom have diabetes and kidney disease, were also confirmed to be carriers of this variant (Figure 3A). The proband’s mother, who does not have diabetes or kidney disease, was not a carrier of the variant. These data demonstrate that the V938Efs*27 variant co-segregates with disease in this family (Figure 3B).
RNASeq Analysis Suggests Alternate Splicing due to c.8300-G>C Variant:
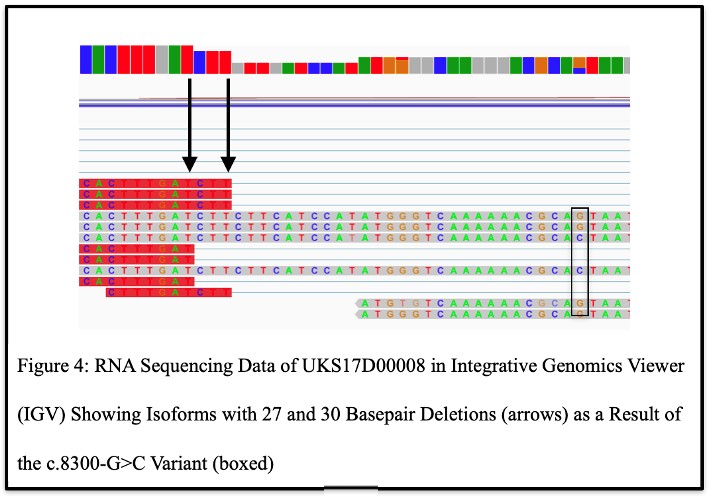
Upon analysis of the RNASeq data from the carrier of C5orf42 pLOF splice variant (c.8300-G>C) with the interactive tool Integrative Genomics Viewer (IGV), two different isomers were discovered, one bearing a 30-bp deletion, and one a 27-bp deletion (Figure 4), suggesting that this variant results in aberrant splicing of C5orf42 mRNA that may lead to potential alternative forms of the CPLANE1 protein.
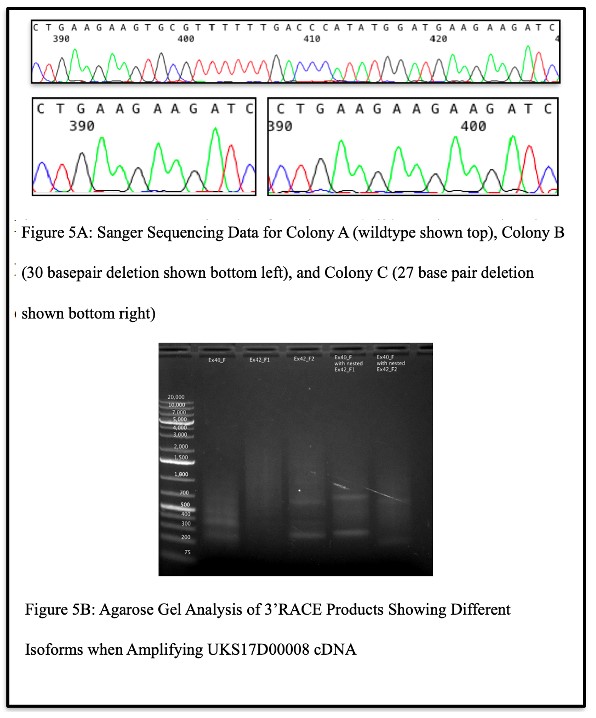
Confirmation of Isoforms:
When attempting to authenticate the observations made above, Sanger sequencing results of DNA isolated from bacterial colonies carrying DNA from patient UKS17D00008 (the carrier of the c.8300-G>C splice variant) showed that some colonies displayed the wildtype sequence, some displayed a sequence with a 30 base pair deletion, and some displayed a sequence with a 27 base pair deletion (Figure 5A). This confirmed observations made with IGV and validated the existence of multiple isoforms caused by the variant. Upon further analysis of the splice variant via the 3’RACE kit, the agarose gel revealed multiple bands when the products were amplified with a gene specific primer in exon 40 of C5orf42, with two gene specific primers in exon 42 of C5orf42, and with nested gene specific primers in exon 40 and 42 (Figure 5B). This suggested the existence of the multiple isoforms and was confirmed by Sanger sequencing the resulting gel
band products, which again revealed the wildtype sequence, a sequence with a 30 base pair deletion, and a sequence with a 27 base pair deletion.
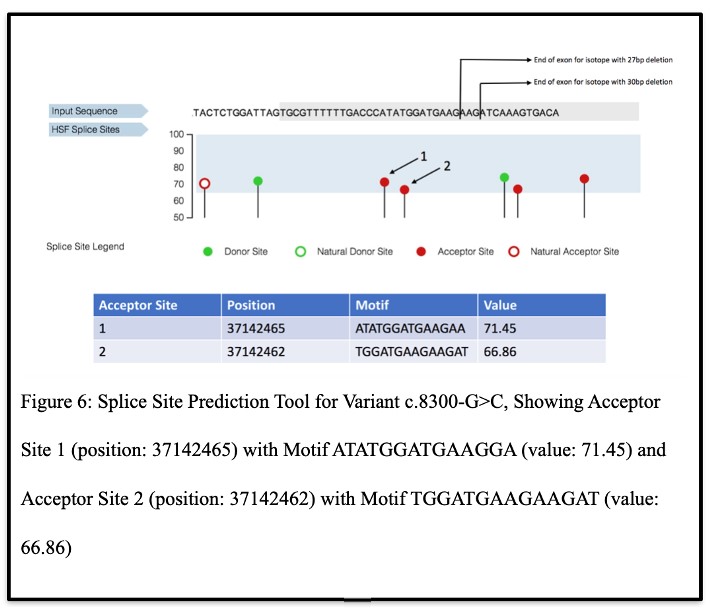
In Silico Confirmation of Alternate Acceptor Sites and C5orf42 Isoforms Due to c.8300-G>C Variant:
To further verify the multiple isoforms, the Genomis HSF Pro system results revealed that variant c.8300-G>C could create two alternate acceptor sites (Figure 6). Each of these sites included in their motifs the isoforms that were discovered through the subcloning and 3’RACE analyses, thereby confirming these results.
DISCUSSION
Through this research, a greater understanding of each variant of C5orf42 identified in participants of the Utah Kidney Study was achieved. Patient UKS17D00022 was determined to be a compound heterozygous carrier of the I165Yfs*17 variant on one allele and the S123F variant on the other. This is important knowledge because this patient could transmit either allele to their children, therefore, causing them to inherit either variant. Its unclear, however, from this study whether one copy or both variants is needed to cause disease. Further research focusing on the rest of this patient’s family and their family’s history with diabetes and DKD may be useful in further analyzing the effects of these variants in C5orf42 on the susceptibility of DKD.
Additionally, the V938Efs*27 variant was found to co-segregate with diabetes and DKD in the family of patient UKS17D00134. This is also important knowledge because it shows a direct relationship between the gene C5orf42 and the susceptibility of DKD. Continuing to analyze descendants of this family could be useful in further supporting this observation and could potentially warrant further genetic testing and genetic counseling of carriers of this variant in this family.
Lastly, two aberrant C5orf42 isoforms were observed as a result of splicing variant c.8300-G>C; these were verified through Sanger sequencing of subcolonies, using 3’RACE amplification, and through in silico prediction using the splice site prediction tool. This discovery is important because it shows that this variant causes alternate splicing of C5orf42 mRNA that results in multiple protein isoforms and may impact protein function. Further research on the functional impact resulting from the 30 base pair deletion and the 27 base pair deletion, which are predicted to result in altered protein isoforms lacking 10 and 9 amino acids, respectively, may be useful in determining how each isoform contributes to the susceptibility of DKD.
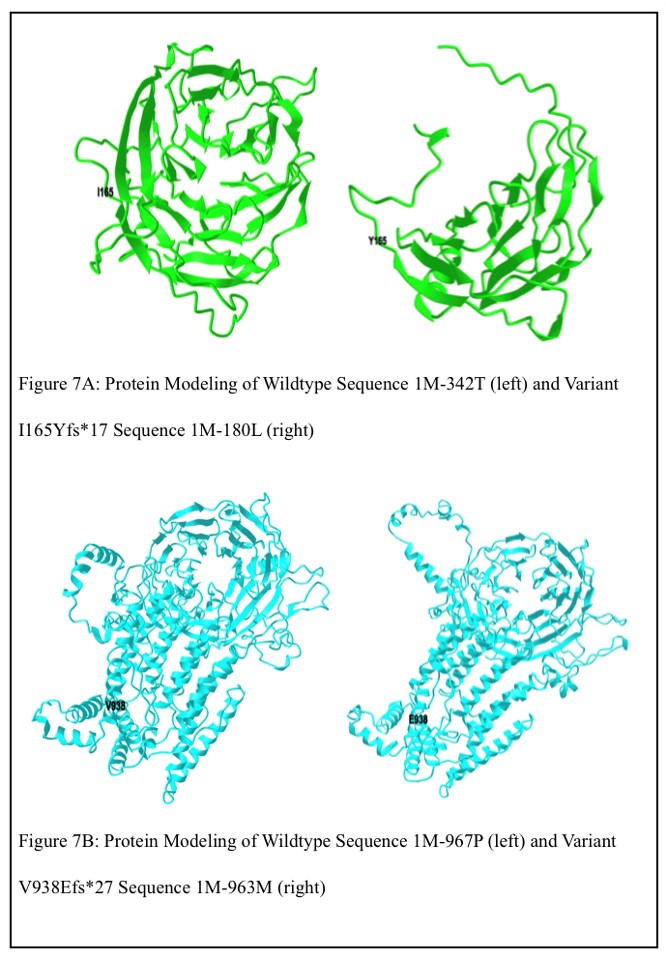
Another interesting finding that further supports the pathogenicity of two of the C5orf42 variants is I165Yfs*17 localizes to the WD40 domain of the CPLANE1 protein and V938Efs*27 localizes near the WD40 domain in the non-cytoplasmic region of the CPLANE1 protein. Many potentially pathogenic variants of proteins in this domain have already been identified and linked to various human pathologies including neurological disorders, cancer, ciliopathies, and endocrine disorders.14 Variants I165Yfs*17 and V938Efs*27 may result in similar effects. With this in mind, the genomic wildtype sequences (chr5:37,106,235-37,107,261 and chr5:37,106,235-37,108,169) and variant sequences of I165Yfs*17 and V938Efs*27 were translated into amino acid sequences using the ExPASy translating tool (https://web.expasy.org/ translate/), and protein modeling of the obtained amino acid wildtype sequences (1M-342T and 1M-967P) and variant sequences of I165Yfs*17 and V938Efs*27 (1M-180L and 1M-963M) was preformed using the Robetta protein structure prediction service (https://robetta.bakerlab.org) to predict the structure of the altered WD40 domain containing proteins relative to the wildtype sequence (Figures 7A and 7B). These differing structures may impact the protein’s function and contribute to each of the patients’ susceptibility to diabetes and DKD.
Our analysis of each of the identified C5orf42 variants further characterized their potential role in DKD and further support the potential benefits of genetic research and how it is important to identify and study possibly deleterious gene variants like those found in C5orf42 in order to diagnose diseases more readily, identify individuals at risk of disease, and possibly identify cures for them in the future.
Additional functional analyses, including in vitro inactivation (knock-out) of C5orf42 and the introduction (knock-in) of each C5orf42 variant, are necessary to further examine the specific phenotypic effects of each variant of C5orf42 and to further investigate the role of this gene in DKD. Transfection with short interfering RNA (siRNA), which binds to target mRNA and mediates its degradation, may be a means for in vitro inactivation and additional investigation of C5orf42’s function. Another gene silencing technique using the clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 system may provide a better understanding of the gene’s function, while also presenting an opportunity for the introduction of each variant of C5orf42. This system involves a guide RNA that matches the target gene along with a Cas 9 (CRISPR- associated) protein that acts as an endonuclease to cause a double-stranded DNA break.15 Once phenotypic effects of each variant are identified in cellular cultures via one of these techniques, proper treatment protocols can be developed in order to help patients who are genetically susceptible to DKD.
REFERENCES
1. Iyengar SK, Sedor JR, Freedman BI, et al. Genome-Wide Association and Trans-ethnic Meta- Analysis for Advanced Diabetic Kidney Disease: Family Investigation of Nephropathy and Diabetes (FIND). PLoS Genet 2015;11:e1005352.
2. McDonough CW, Palmer ND, Hicks PJ, et al. A genome-wide association study for diabetic nephropathy genes in African Americans. Kidney Int 2011;79:563-72.
3. Pezzolesi MG, Poznik GD, Mychaleckyj JC, et al. Genome-wide association scan for diabetic nephropathy susceptibility genes in type 1 diabetes. Diabetes 2009;58:1403-10.
4. Sandholm N, Forsblom C, Makinen VP, et al. Genome-wide association study of urinary albumin excretion rate in patients with type 1 diabetes. Diabetologia 2014;57:1143-53.
5. Sandholm N, Salem RM, McKnight AJ, et al. New susceptibility loci associated with kidney disease in type 1 diabetes. PLoS Genet 2012;8:e1002921.
6. Sandholm N, Van Zuydam N, Ahlqvist E, et al. The Genetic Landscape of Renal Complications in Type 1 Diabetes. J Am Soc Nephrol 2017;28:557-74.
- Lazaro-Guevara J, Fierro-Morales J, Wright AH, et al. Targeted Next-Generation Sequencing Identifies Pathogenic Variants in Diabetic Kidney Disease. Am J Nephrol 2021;52:239-49.
- Romani M, Mancini F, Micalizzi A, et al. Oral-facial-digital syndrome type VI: is C5orf42
really the major gene? Hum Genet 2015;134:123-6.
- Wentzensen IM, Johnston JJ, Keppler-Noreuil K, et al. Exome sequencing identifies novel mutations in C5orf42 in patients with Joubert syndrome with oral-facial-digital anomalies. Hum Genome Var 2015;2:15045.
- Fleming LR, Doherty DA, Parisi MA, et al. Prospective Evaluation of Kidney Disease in Joubert Syndrome. Clin J Am Soc Nephrol 2017;12:1962-73.
- Ma RC, Cooper ME. Genetics of Diabetic Kidney Disease-From the Worst of Nightmares to the Light of Dawn? J Am Soc Nephrol 2017;28:389-93.
- Sas KM, Yin H, Fitzgibbon WR, et al. Hyperglycemia in the absence of cilia accelerates cystogenesis and induces renal damage. Am J Physiol Renal Physiol 2015;309:F79-87.
- Drivas TG, Lucas A, Zhang X, Ritchie MD. Mendelian pathway analysis of laboratory traits reveals distinct roles for ciliary subcompartments in common disease pathogenesis. Am J Hum Genet 2021;108:482-501.
- Yeonjoo K, Soo-Hyun K. WD40-Repeat Proteins in Ciliopathies and Congenital Disorders of Endocrine System. EnM Endocrinol Metab 2020; 302:1-13.
- Redman M, King A, Watson C, King D. What is CRISPR/Cas9. et al. Arch Dis Child EducPract Ed 2016; 101:213–215.