John and Marcia Price College of Engineering
12 Computational Investigation of Ammonia/Hydrogen Combustion in Premixed Flames
Joseph Lee and Alex Novoselov
Faculty Mentor: Alex Novoselov (Mechanical Engineering, University of Utah)
Introduction
In recent decades, there has been a significant increase in interest of carbon-free fuels due to rising energy demands across the globe and their contributions to climate change. One such potential alternative is hydrogen (H2). However, due to its high mass-diffusivity, fractal flame propagation is observed at conditions relevant to practical devices. A promising solution is the inclusion of ammonia (NH3) in a hydrogen combustion system. Ammonia, also a carbon-free fuel, does not propagate in a fractal nature and can be used as both a vector for stabilization and storage of hydrogen.
Combustion systems are difficult to study physically due to their turbulent high-temperature nature, thus computational systems become a primary tool for investigating such systems. Many chemical mechanisms have been developed to try and model hydrogen/ammonia combustion, but each one is often tailored to specific conditions. To choose an appropriate mechanism from this variety of mechanisms, accuracy of said mechanisms must be validated against tangible experimental data.
The mechanisms selected in this work are based off previous analysis by Alnasif et al. [1]. Among the top performing mechanisms in Alnasif’s study are those developed by Duynslaegher [2], Glarborg [3], Gotama [4], Lamoureux [5], Nakamura [6], and UC San Diego (with nitrogen chemistry) [7]. The Glarborg mechanism focuses on nitrogen/oxygen chemistry due to its focus on nitrous oxides (NOx), with carbon species included to simulate carbon-scrubbing mechanisms [3]. Duynslaegher, being an older mechanism from 2012, focused on updating reaction rate constants and improving NH2 and N2O simulation pathways with novel results from literature [2]. Lamoureux focuses on prompt-NO formation in low-pressure flames [5], Nakamura looks at weak flames [6], and Gotama investigates fuel-rich high-pressure systems and optimizes the chemical mechanism from Han et al. [4] [8], while the mechanism developed at U.C. San Diego is tailored towards high temperature ignition and detonations [7]. These mechanisms will be referred to by first author and year of publication henceforth. This work is a preliminary investigation into a wide variety of conditions investigated by Lhuillier et al. [9], with results informing further investigations
Methods
To evaluate the accuracy of said mechanisms, simulations of one dimensional adiabatic freely propagating premixed flames were performed using the FreeFlame mode in Cantera [10] with varying ammonia/hydrogen mixtures as fuel. The laminar flame speed is observed for varying conditions corresponding to experimental conditions investigated by Lhuillier et al. [9]. All simulations are done at an unburned atmospheric pressure of 1 atm. Unburned temperatures of 298 K and 473 K were chosen to be investigated, giving insight into physical situations like flame propagation from room temperature (298 K) or engine preheat conditions (473 K). Equivalence ratios investigated in this work cover ranges of Φ = 0.8-1.4. Ammonia/hydrogen ratios (by volume content) of 100%/0% (pure air), 70%/30%, and 40%/60% are investigated to give an overview of behavior over a wide range for laminar flame speeds (sL) produced by each mechanism. A standardized grid of 1000 points across was used in favor of adaptive mesh refinement (AMR) to enable more accurate comparison between flames. To evaluate accuracy, absolute error between simulated results and experimental results was analyzed using
[latex]\begin{equation} \% \; error = \left| \frac{s_{L,sim} - s_{L,exp}}{s_{L,exp}} \right| \times 100 \end{equation}[/latex]
where sL,sim represents the flame speed calculated, and sL,exp represents the experimental data as measured by Lhuillier [9]. These errors were then integrated over all conditions (equivalence ration and ammonia/hydrogen ratio) for comparison. A ranking is determined from these results to help inform users of their choice of chemical mechanisms to use.
Results
As seen in Figure 1, the chemical mechanism used to perform combustion simulations greatly affects outcomes. Here, the predicted flame speed supersedes experimental values on average. Trends, such as larger error presented with the Lamoureux 2016 mechanism, are expected due to their tailoring to conditions not investigated in this work.
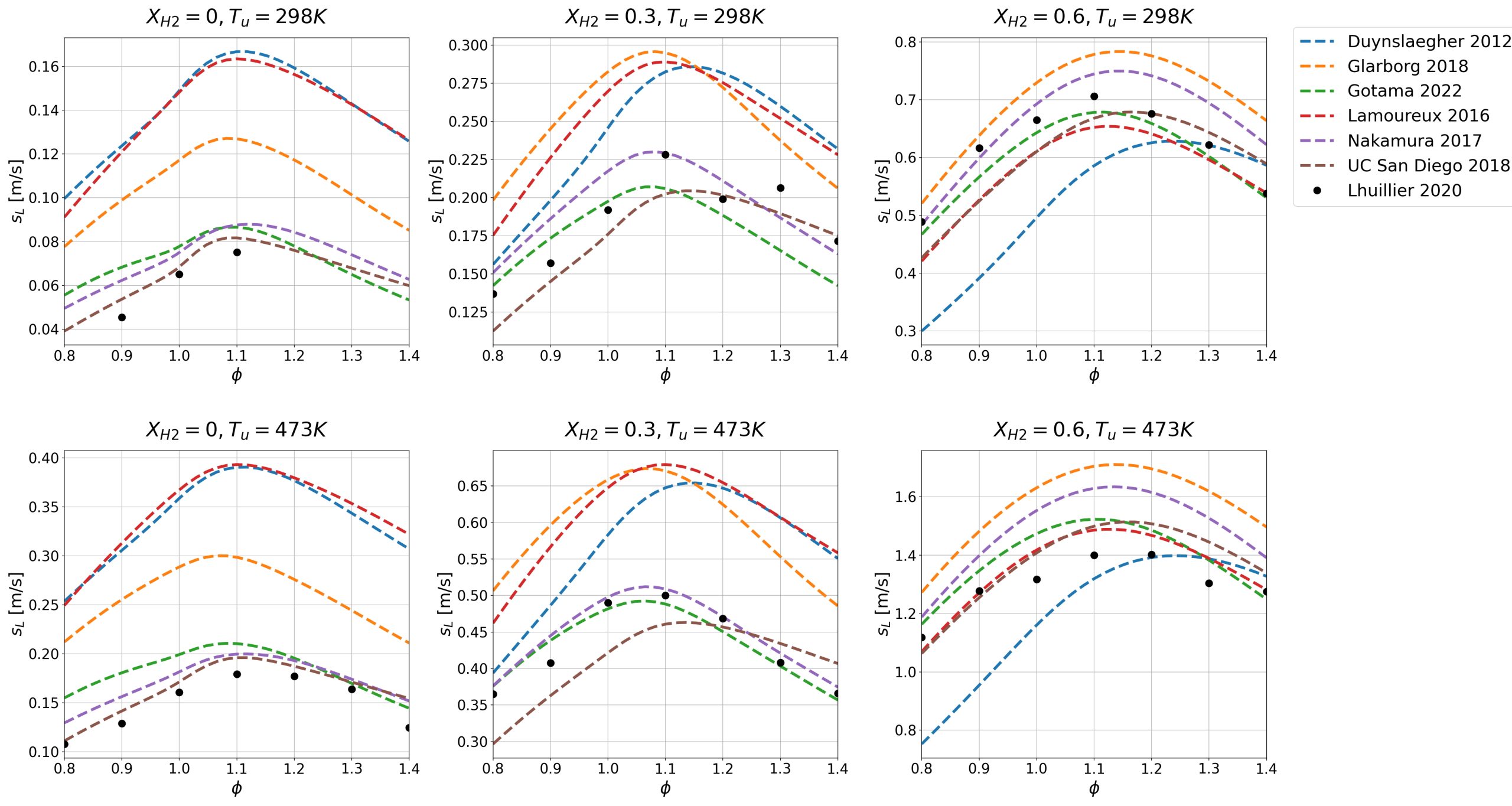
Figure 1: Laminar burning velocities of investigated chemical mechanisms at various NH3/H2 mixtures at unburned pressures of 1 atm, compared against experimental data from Lhuillier et al. [9] (black points).
From analysis of integrated errors on the laminar flame speed (sL), the most accurate mechanism at low and high unburned temperatures is the one developed at U.C. San Diego. From Figure 2, we can see that the rankings for accuracy at predicting laminar flame speeds at 298 K are:
The rankings for accuracy at predicting laminar flame speeds at 473 K are:

The trend of newer mechanisms being more accurate on average is expected due to the fact that succeeding works should seek to improve upon older literature.
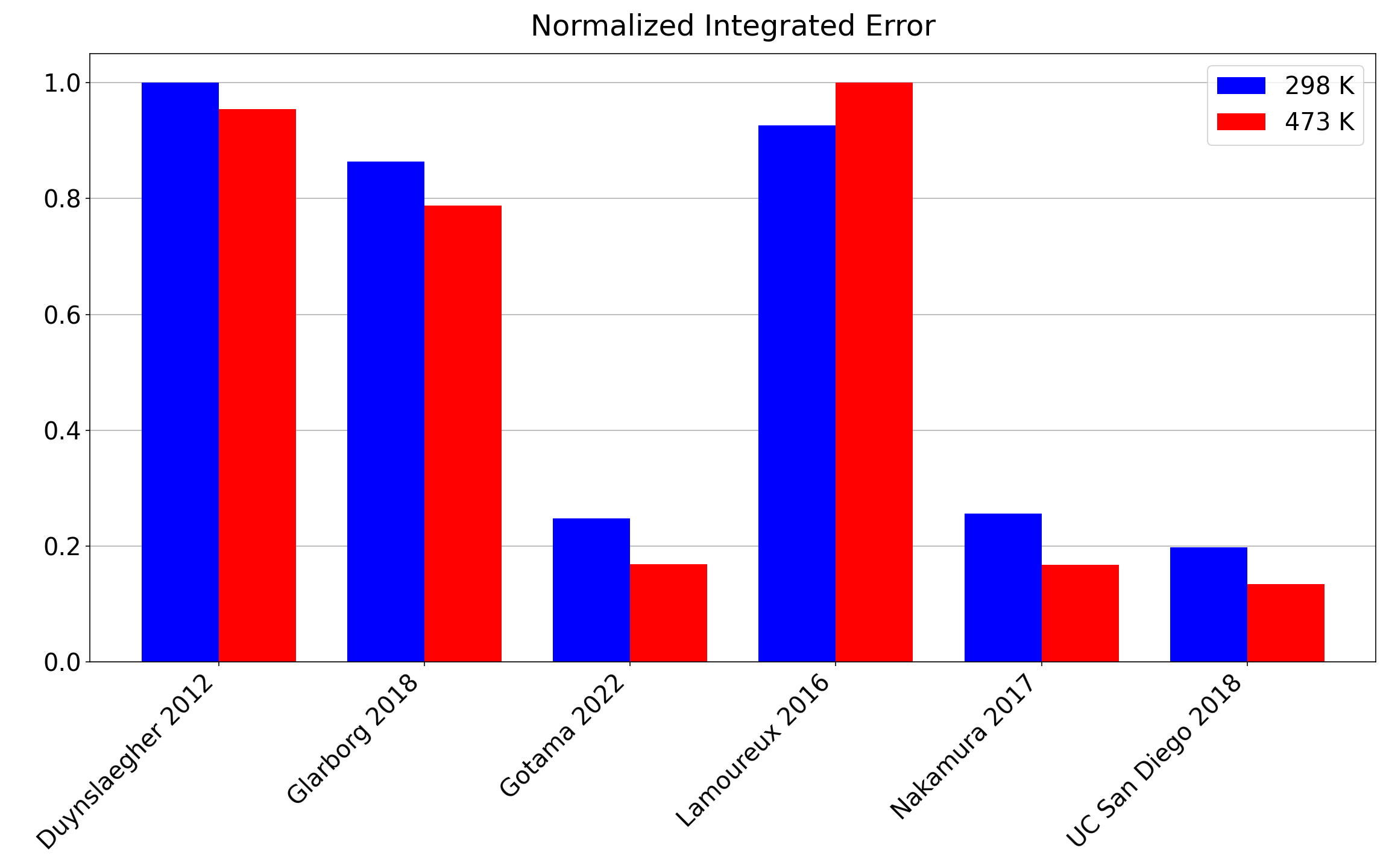
Figure 2: Normalized integrated errors of each chemical mechanism over all equivalence ratios (ϕ) at 298 K (blue) and 473 K (red).
Conclusions
Overall, the U.C. San Diego 2018 mechanism exhibits the least error in laminar flame speed, closely followed by the Nakamura 2017 and Gotama 2022 mechanisms. It should be noted that while mechanisms like Duynslaegher 2012 and Lamoureux 2016 have higher errors associated with them, these mechanisms also tend to have a shorter runtime. Large simulations, such as full three-dimensional simulations performed by national laboratories [11], may prefer less accurate but faster mechanisms due to associated computational costs [12].
The investigations performed in this work point to drastic differences between chemical mechanisms in combustion due to each being tailored to very specific conditions. This warrants further investigation over a broader range of chemical mechanisms, as well as more conditions. The research phase of this work also unveiled large runtime differences between mechanisms. Further work is to be done comparing accuracy to runtimes to better inform users, both of which are factors to consider when running computational models.
Acknowledgements
This work was supported by UROP from the Office of Undergraduate Research at the University of Utah awarded to Joey Lee. The support and resources from the Center for High Performance Computing at the University of Utah are gratefully acknowledged.
Footnotes
Media Attributions
- 137489262_sl
- rankings1_298K
- rankings2_473K
- 146879582_error
