John and Marcia Price College of Engineering
30 Protein Z as a Potential Anti-Fibrotic Therapeutic for Cardiovascular Fibrosis
Georgiy Polishchuk; Thirupura Shankar; and Stavros Drakos
Mentor: Thirupura Shankar (Biomedical Engineering, University of Utah)
Abstract
No effective non-invasive treatments exist for heart failure (HF). This paper aims to determine whether Protein Z may function as a potential antifibrotic target to develop noninvasive and preventative treatments for HF. An acute hypertensive mouse model and human cardiac fibroblast (HCF) culture were employed to observe Protein Z expression’s effect on fibroblast activity and consequent fibrosis development. Protein Z expression was manipulated within HCF samples following stimulation to induce over expression (OE) or knockdown (KD). Similarly, mice, excluding the saline control, received a treatment to induce hypertension for 4 weeks with one group receiving a viral injection to induce KD. Echocardiography was performed to observe mouse heart function. At the conclusion, immunohistochemistry, tissue weighing, and protein quantification was performed to observe fibroblast activity and fibrosis development. The results demonstrated that decreased Protein Z expression led to decreased fibroblast activity and protein excretion. Similarly, tissue imaging suggested improved tissue structure following Protein Z downregulation. Thus, this study demonstrated Protein Z’s potential as an antifibrotic target. Heart failure is intricately tied to fibrosis development and adverse cardiac remodeling. Antifibrotic targets are promising therapeutics to limit adverse cardiac remodeling and improve heart function.
Index Terms— Cardiovascular Illness, Fibroblast, Fibrosis, Gene Expression, Hypertension
Introduction
Cardiovascular diseases (CVDs) are the leading causes of death globally and account for 34% of deaths in the United States. In 2006 alone, CVDs accounted for over $400 billion in health care spending [1]. Following cardiac stress, such as inflammation or myocardial infarction (MI), the over- activation of fibroblasts can lead to excessive extracellular matrix (ECM) deposition, termed replacement fibrosis [2]. Increased myocardial fibrosis is one of the main drivers of systolic heart failure (HF), which limits the heart’s ability to pump blood adequately to the rest of the organs [3]. No effective non-invasive methods exist for treating replacement fibrosis, indicating the need to regulate fibroblast activity. Although the initial process of ECM excretion is necessary for recovery following heart damage, the over-deposition of ECM can deteriorate heart function. When cardiomyocytes die, the space left by the cells must be filled with ECM to preserve heart structure and function. In adverse remodeling, fibroblasts over deposit ECM, causing heart tissue to stiffen and lose contractility [2]. Unfortunately, adverse remodeling can lead to heart failure (HF) with complete degradation of heart function. Thus, promising treatments for fibrosis center around restricting fibroblast activity, a vital pathway in fibrosis development [3]. Fibroblasts serve the function of excreting ECM to maintain structure following tissue damage but require differentiation into myofibroblasts to actively carry out this process. Protein Z could potentially regulate the development of fibrosis through its role as a transcription factor [6] [11]. Successful control over fibroblast activation may improve HF patient outcomes without invasive operations.
Current research investigating fibrosis centers around modulating fibroblast gene expression to limit fibrosis. Various proteins regulate fibroblasts through gene regulation as transcription factors [9]. Thus. controlling upstream genes serves as a potentially effective therapeutic method. However, the mechanism behind fibrosis is not fully understood, as various cells can differentiate into fibroblasts under unique conditions and biochemical signals [9]. Although limited information is known about the mechanism behind Protein Z- mediated cardiac fibrosis, recent studies have promising findings about the use of Protein Z in halting pulmonary, hepatic, and renal fibrosis [6]. However, no conclusive research has been conducted to understand Protein Z’s role in cardiovascular fibrosis.
Exploring Protein Z’s function as a transcription factor related to fibroblast differentiation is vital in understanding the mechanism behind Protein Z-mediated cardiovascular fibrosis. Thus, this paper aims to investigate the role of Protein Z inhibition in preventing excess cardiac fibrosis. Moreover, additional research could reveal its potential as a therapeutic target for cardiac fibrosis. In this work, Protein Z expression was manipulated using adenovirus on human cardiac fibroblasts (HCF) to investigate whether Protein Z causes fibroblast activation. Additionally, an acute hypertension induced heart failure model was employed to observe the effect of Protein Z expression on regulating adverse cardiac remodeling. Showcasing Protein Z’s role in mediating cardiac fibrosis will determine whether the protein merits additional investigation into its mechanism and as a potential therapeutic target.
Background
Fibrosis Developmentand Hypertensive Model
As excess fibrosis is a common cause of HF, many studies explore therapeutic targets to limit fibrosis and adverse cardiac remodeling. Specifically, studies center around evaluating interstitial and replacement fibrosis due to the condition’s effect on adverse remodeling. Interstitial fibrosis particularly can push cells like cardiomyocytes out of alignment [2]. which adversely affects tissue function. Beyond HF, fibrosis is associated with many cardiovascular complications and conditions. As human heart tissue degenerates and deteriorates, the contractile force gradually decreases. Fibrosis can worsen cardiovascular conditions like hypertension with the occlusion of blood vessels through ECM growth [12]. Conversely, hypertension alters the heart’s workload and causes the overworked left ventricle to enlarge. The excretion of ECM by the heart because of hypertension can then progress to unfavorable remodeling and heart failure [13].
Fibrosis Marker sand Key Proteins
While the mechanisms behind fibrosis development are varied and complex, exploring the regulation of transcription factors like Protein Z can reveal potential therapeutics in mitigating and potentially reversing adverse remodeling. Studies show that Protein Z activates fibroblasts by regulating protein expression [6]. Specifically, Protein Z is essential in the MAPK and NF-kB signaling pathways, which promote the differentiation of fibroblasts [6]. Additionally, our preliminary studies using adenovirus (AdV-CMV-h Protein Z -GFP) on human cardiac fibroblasts (HCF) resulted in increased expression of smooth muscle actin (SMA), collagen, and smooth muscle-specific protein (SM22), evidence for the activation of fibroblasts [16]. SM22 and SMA are markers of active fibroblast differentiation and myofibroblasts, respectively [7]. Importantly, collagen comprises a significant portion of the ECM and consequently serves as a marker of fibrosis maturation [4].
Certain proteins serve as markers of fibroblast development and maturation. For instance, factors like TGFβ mediate the differentiation of certain cell types into fibroblasts. While there are multiple pathways in which cells differentiate into myofibroblasts, TGFβ is an important growth factor which allows for adipocytes, fibroblasts, endothelial, and epithelial cells to differentiate [11]. Therefore, quantifying these proteins within experimental models reveals ECM remodeling of the heart and progression of fibrosis.
HF Mechanisms
As the mechanism for the development of fibrosis lacks definitive consensus, various treatments center around patients already experiencing HF through reactive rather than preventative care. Many pathways lead to the development of fibrosis, such as the RAAS pathway [14]. As such, many proposed potential anti-fibrotic therapeutics center around regulating connected biological systems such as the RAAS pathway [14]. However, therapeutics aimed at modulation of fibroblast activity prior to extreme cardiac remodeling hope to provide preventive measures against HF or eventual transplantation. Greater understanding of Protein Z’s role in the development of fibrosis will provide insight into its potential as a therapeutic target against fibrosis and a preventative medicine against HF.
Experimental Models
Considering the various models to employ in studying cardiac fibrosis is essential to ensure the study is medically applicable. Many anti-fibrotic therapeutic studies fail due to the use of inappropriate animal models and lack of cell specificity [4]. Animal models alone fail to capture the complexities behind human fibrosis, which tends to be chronic rather than acute [4]. Within humans, the development of heart failure can take decades to fully manifest with multiple factors influencing the severity of the condition and ratio of interstitial versus replacement fibrosis [2]. In contrast, heart failure can be induced within weeks for animal models which allows for practical experimental observation but fails to capture the nuance behind human fibrosis. Employing a mouse model using hypertension can allow for an acute study of gradual fibrosis development as hypertension puts strain on the heart [2]. A hypertensive model can be effectively used, as the excessive load on the heart’s muscle fibers leads to oxidative stress and potential cell death [15]. Further research performed by the Drakos’ Lab later employed a chronic mouse model and human tissue culture to study Protein Z’s potential as a fibrotic therapeutic target. The lab is now working to fully understand the mechanism behind Protein Z mediated cardiac fibrosis.
METHODS
As the study aims to determine the potential role of Protein Z in regulating the progression of cardiac fibrosis, Protein Z expression was manipulated using adenovirus. Following stimulation, differentiated myofibroblasts were treated with a viral injection to observe differences in Protein Z overexpression (OE) and knockdown (KD). Similarly, an acute hypertensive mouse model was used with an experimental KD group with a positive and negative control group. Target proteins reflective of fibroblast differentiation and ECM expression were quantified through Western Blotting and differences were statistically analyzed. Additional histology was performed with the mouse model to evaluate heart structure, specifically with replacement and interstitial fibrosis. Evaluating differences between experimental groups and controls demonstrated the role of Protein Z expression in the progression of cardiac fibrosis.
HCF Cell Culture
HCF from PromoCell, #c-12375, were cultured in 12-well plates with 50,000 cells per well with PromoCell HCF media, #C-23010, for 48 hours. The cells within each well were stimulated by 10 ng/ml of TGFβ for 48 hours during which media change occurred every 24 hours. 72 hours following or preceding stimulation, a viral infection was performed at 250 multiplicities of infection for 24 hours. Both Protein-Z OE with AD- CMV-h Protein-Z -GFP and KD with Ad-CMV-sh- Protein-Z -GFP were achieved. Each injected vector was in a concentration of vector in a concentration of 3.1* 10e12 VG kg−1 with the GFP vector serving as a control. Upon conclusion of the experiment, cells were harvested with a cell scraper. Molecular characterizations were then performed.
HCF Extraction
HCF were lysed in 1X Cell Signaling Technology RIPA buffer, #9806S, with 2x protease and phosphatase inhibitor (Thermo Scientific #78440). The cells then sat in solution for 20 minutes, after which 5μl of 100 mmol phenylmethylsulfonyl fluoride was added to solution to inhibit proteases. The homogenate rotated at 4°C for 30 minutes before centrifugation at 4°C at 18407 rcf. The supernatant was transferred into a new tube for protein estimation using Pierce BCA Protein Assay kit (Thermo Scientific, #23225). 2x Laemmli buffer was introduced to the supernatant in equal part to 10% DTT. The solution was boiled for 10 minutes at 98°C. A similar process occurred for mouse left ventricular (LV) tissue samples except that a 30 μg sample of transmural LV was first homogenized using metal beads for 3 min within the previously described solution.
Animal Care
The animal study with the acute mouse model was performed in accordance with the Institutional Animal Care and Use Committee (IACUC). Research procedures such as surgical operations, treatment administration, and observation were approved by the Animal Care and Use Committee of the University of Utah and complied with the American Physiological Society’s Guiding Principles in the Care and Use of Animals and the UK Animals (Scientific Procedures) Act 1986 guidelines. In accordance with University of Utah protocol, the mice were housed in the intuition at 70 °F and 40% humidity with 12 h dark/light cycle. All groups received the same diet and care conditions with the only variance being in specific treatment administered.
Angiotens in II and Phenylephrine (AngII+PE)Systemic Fibrosis Model:
Male and female 12-week-old C57BL6J mice were used within the study which had systemic fibrosis induced through continuous infusion of AngII+PE for two weeks. Infusion was regulated by an Alzet mini-osmotic pump (model #2004) which was prepared as per manufacturers protocol with saline or a combination of Angiotensin II (1.5mg/g/day) and Phenylephrine (50mg/g/day). Two weeks following introduction of the pump, mice randomly received an injection of AAV9-CMV-shRNA-m-Protein-Z-GFP vector or control AAV9-CMV-GFP vector in a concentration of 3.1* 10e12 VG kg−1. The three groups consisted of 5 mice (N=5) and were recurrently echoed for entire 4-week duration of study, notably 2-weeks prior and following injection. Saline mice only had a saline pump introduced, the GFP group had the control vector, while the Protein-Z group had the experimental vector introduced. The pumps were weighed at the start and end of the study. Upon harvesting, the hearts were washed with 1x Phosphate Buffered Saline Solution (PBS). Upon completion of the study mouse hearts were weighed, Immunohistochemistry (IHC) was performed, and molecular characterization was performed.
Echocardiogram
Throughout the four-week timeframe of the systemic fibrosis model, mice were weekly observed through ECG using limb leads. The mice were anesthetized with 1.5% isoflurane and .8 liters of oxygen during. While under anesthesia, images of the mice heart and aorta were taken on the Vivo system along the 2D long and short-axis. Analysis of the images was performed in Vivo strain software (version 3.1.1). Echocardiograms were performed serially over 2 consecutive cardiac cycles to conduct analysis of the Ejectile Fraction (EF), Left Ventricular End Diastolic Diameter (LVEDD), and Fractional Shortening(FS).
Western Blotting
30 μg of protein were pipetted into polyacrylamide gel which were run at a constant 25V per gel until the ladder was around the bottom of gel.. The proteins were then transferred onto a nitrocellulose membrane at a constant 350 mA for 1 hour. A total protein stain (TPS) was performed before being scanned following the Licor Revert 700 Total Protein Stain protocol. The membranes were blocked with Odyssey Blocking Buffer (LiCor #927-50000) and probed with primary antibodies overnight in a cool room and constant motion to promote mixing. The blots were then washed with 1x tris- buffered saline with tween (TBS-tween) three times. Following the wash, the protocol was performed in the dark. The blots were incubated with secondary antibodies (anti- mouse or anti-rabbit 1:10,000) for 30 mins and washed an additional three times. The blots were then scanned with Odyssey Infrared Imager (LI-COR 9120). Each gel had a unique TPS which allowed for present proteins to be normalized, and the blots were analyzed with LI-COR Image Studio Lite software version 5.2.
IHC
Mice tissue, kidney, cardiac, or other tissues of interest, were embedded in OCT solution (Sakura Finetek) after PFA fixation in 4% paraformaldehyde. The samples were then frozen and stored at -80ºC prior to sectioning with the cryostat (Leica CM1950). The tissue was sectioned around 100μm thickness before being stained in 4’,6-diamidino-2- phenylindole (DAPI) (ThermoFisher Scientific D3571) at a concentration of 1:1000, Wheat Germ Agglutinin (WGA (ThermoFisher Scientific W32464) at a concentration of 1:1000, Periostin/OSF-2 Antibody (Novus Biologicals 30042) at a concentration of 1:100, and Protein Z (Santa Cruz Biotechnology) at a concentration of 1:10. Anti-mouse and anti-rabbit secondary antibodies were used accordingly. The images were taken using Leica Application Suite X (3.5.7.23225) with identical laser settings across each sample and a z-depth within 20 µm of each other. Images for each protein of interest were acquired with the same laser settings of a confocal microscope (Leica SP8, Leica Microsystems CMS GmbH, Leica DMi8 automated) across samples and processed using Fiji (ImageJ 2.3.0). Tissue structure was then visually evaluated with special attention to areas of concentrated ECM. Interstitial, replacement fibrosis, and cardiomyocyte orientation were evaluated between each of the three groups.
Statistical Analysis
Protein quantification data from Western Blotting was compared across different experimental groups. The relative concentrations of target proteins were evaluated as the mean ± standard error of the mean (SEM). The analytical and data visualization software, GraphPad Prism (version8.2.1, GraphPad), was employed for all statistical analyses. Two- tailed tests were performed in comparisons of the cell culture experimental groups. In the mouse study, one-way ANOVA was used for comparing multiple groups. A p-value < 0.05 was considered statistically significant. All experiments were repeated independently with at least three biological replicates. Western blot quantification was performed from the same gel or processed in parallel when necessary.
RESULTS
Following stimulation of the fibroblasts in the cell culture, the cells were treated such that there was a Protein-Z OE and KD experimental groups. The cells were lysed, and Western Blotting was performed to quantify relative amounts of target proteins. The KD group excreted less collagen than the stimulated group while the OE excreted four times as much collagen (Figure 1).
Similarly, markers of fibroblast differentiation and activity such as Sm22 and SMA were in significantly higher concentrations in the OE group compared to the stimulated group. The OE group demonstrated increased activity of fibroblasts in vitro (Figure 2). The KD group conversely had significantly lower levels of Sm22 and Collagen compared to the unstimulated group (Figure 3). Notably, the KD group excreted five times less collagen than the GFP group.
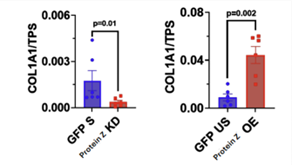
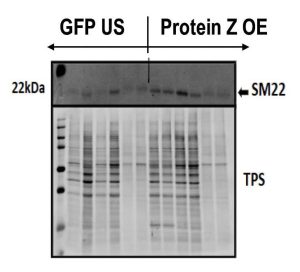
Figure 1: Representative Western Blott Image of a collagen stain for the KD group. Quantification of Protein Z (KD) of Collagen (n=6) and Protein Z Overexpression (OE) from Western Blott data showcasing relationship of Protein Z expression to ECM deposition.
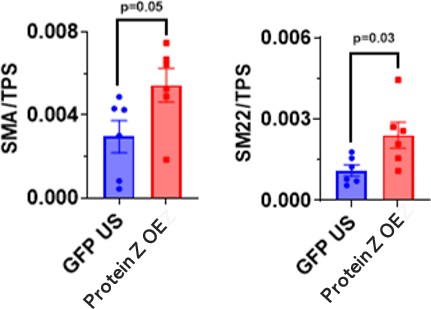
Figure 2: Western blot quantification of HCF culture. The proteins SMA and SM22 in unstimulated (US) and Protein Z OE (n=5 each) are displayed. P- value derived from Two-tailed unpaired t-test.
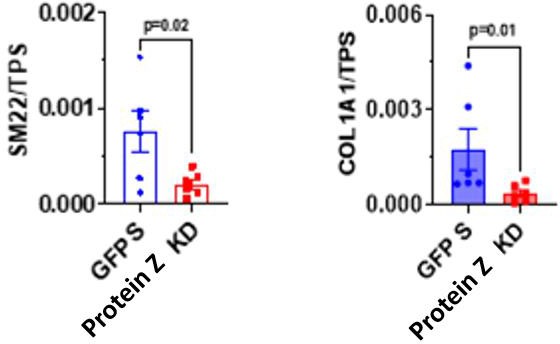
Figure 3: Western blot quantification of HCF culture. The proteins SMA and SM22 in unstimulated (US) and Protein Z OE (n=5 each) are displayed. P- value derived from Two-tailed unpaired t-test.
The mouse study was conducted to model systemic fibrosis through hypertension, over the 4-week duration of which echocardiogram and ECG observation and analysis were conducted, shown in Figure 4. Both the KD and GFP groups had thicker left ventricular walls compared to the saline group towards the end of the study. The GFP and KD groups started with similar ejectile fractions (EF) of the left ventricle. Towards the conclusion of the study, no statistically significant differences were observed between with the EF; however, the GFP and KD groups seemed to change from the saline control towards the end of the study.
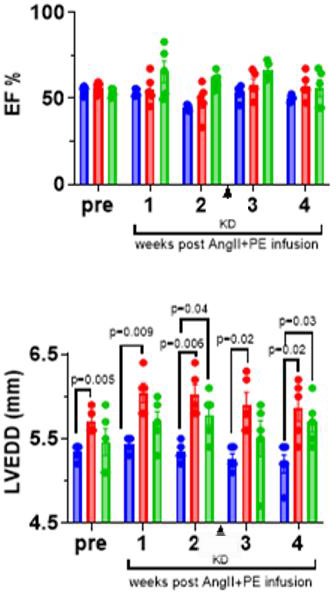
Figure 4: Longitudinal echocardiography data showing left ventricular ejection fraction (EF%) and end-diastolic diameter (LVEDD) respectively (n=5 each). p-value: Multiple two-tailed t-test.
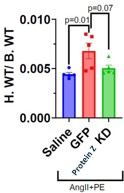
Figure 5: Mouse heart mass/body mass of systemic fibrosis model (n=5)
Upon harvesting the hearts of the system fibrosis models, the hearts were weighed, and Western Blotting was performed. The GFP group had significantly heavier hearts compared to the Saline group. The Protein Z KD group, while not significant, had around 20% lower average heart weight compared to the GFP group (Figure 5).
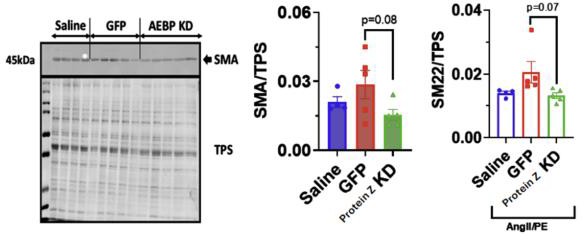
Figure 6: Representative Western Blott image of systemic model SMA stain. Quantification of SM22 (n=4 saline, n=5 GFP and Protein Z KD). Quantification of SMA (n=4 saline, n=5 GFP and Protein Z KD).
Quantified Western Blott data, seen in Figure 6, of the systemic model did not demonstrate significant differences in concentrations of SMA or SM22 between the GFP and Protein Z KD groups. However, the Protein Z KD groups had on average fewer relative amounts of SMA and SM22 compared to the GFP group.
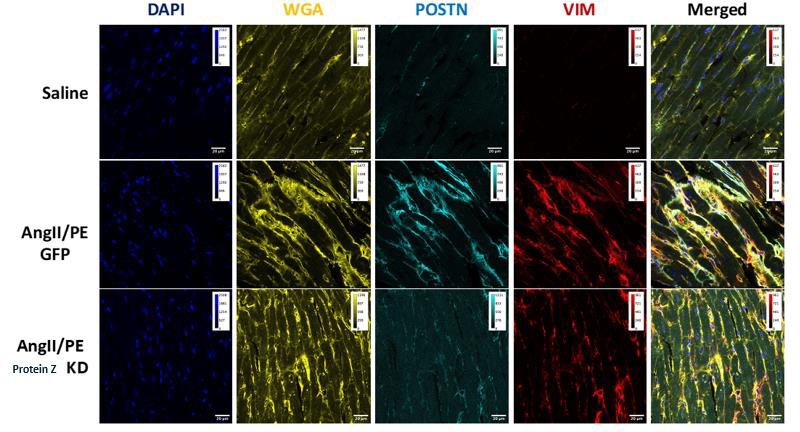
Figure 7: Immunohistochemistry (IHC) imaging of mouse myocardium (n=5 each)
IHC data in Figure 7 showed increased ECM proteins and more irregular tissue structure in the GFP group compared to both the Saline and Protein Z KD groups. The Saline group witnessed less ECM and demonstrated a baseline structure of heart tissue.
Discussion
Cardiovascular illness continues to be the leading cause of death in the US with heart failure being a large contributing condition [1]. With fibrosis functioning as a main driver of heart failure, therapeutics limiting the development of fibrosis may serve as preventative measures against death from cardiovascular illness [3]. Accordingly, Protein Z has been tied to the development of fibrosis within other organ systems and may serve as a potential antifibrotic therapeutic [6, 11]. Unfortunately, the development of cardiac fibrosis as mediated by Protein Z is not well understood. Accordingly, this study aims to explore the effect of Protein Z expression on the development of cardiac fibrosis by employing a human cardiac fibroblast (HCF) cell culture model and an acute hypertension mouse model.
Results from the HCF culture demonstrated an increased fibroblast activity associated with Protein Z expression. Following stimulation, activated fibroblasts demonstrated significantly increased levels of key proteins secreted by activated fibroblasts such as SM22 and SMA (Figure 3). Protein Z KD prevented cardiac fibroblast activation and reduced ECM deposition in vitro. Moreover, HCF cells that expressed Protein Z were shown to secrete significantly more collagen (Figure 2). Such results showcased Protein Z’s role in the development of fibrosis: HCF increased activity and secreted more ECM with Protein Z expression. The observed effect of Protein-Z expression and fibroblast activity seems to confirm previous studies observation of Protein Z as a transcription factor [11].
Similarly, results from the mouse model study demonstrated differences in cardiac structure following Protein Z expression. While no statistically significant differences were observed between the positive control group and the KD group’s cardiovascular function, as the study progressed, echocardiogram LVEDD data seemed to suggest emerging differences between the groups (Figure 4). Such observations were reflected within the average heart weight, as the KD group had smaller hearts on average, albeit, not with statistical significance (Figure 5). The observations suggest that limiting activity of Protein Z expression may lead to decreased fibrosis due to limited fibroblast activity. Similar findings were reported within pulmonary and renal fibrosis studies where limiting Protein Z expression resulted in less fibrosis development [6,9]. While no studies have observed the connection between fibrosis and Protein Z expression, our study demonstrates a potential pathway of fibrosis development mediated by Protein Z.
IHC data from the mouse models was analyzed for structural differences between the groups. The KD group appeared to have a more regular structure witnessed the cardiomyocytes lining the tissue in parallel with one another (Figure 6). Additionally, the WGA staining illuminated increased regions of fibrotic tissue within both the KD and positive control groups demonstrating both groups undergoing the scarring process following cardiac injury. However, the KD group appeared to have less overall fibrosis with less widespread areas of replacement and interstitial fibrosis. These results offer insight into Protein Z as a potential antifibrotic target and as a preventative treatment to current LVAD and heart transplant treatments for heart failure [3]. Our study offers insight into treating heart failure without invasive procedures by investigating therapeutic targets that offer the heart a chance to recover following damage but avoid adverse cardiac remodeling through fibrosis.
The study showed the potential behind downregulating Protein Z expression to limit the development of fibrosis. However, additional work is required to fully understand the mechanism behind Protein Z-mediated fibrosis. Specifically, further research should elucidate what proteins and genes Protein Z interacts with that may indirectly influence the progression of fibrosis. Full comprehension of the Protein Z mechanism may offer insight into additional methods to produce effective anti-fibrotic therapeutics and their side effects.
This study was not without limitations. While demonstrating a link between the development of cardiac fibrosis and Protein Z expression, the study did not define the mechanism behind Protein Z-mediated cardiac fibrosis. The use of an acute mouse model limited the study’s generalizability to human heart failure, which occurs over longer durations of time. Future work by the Drakos’ lab aims to address said limitations. Employing a chronic heart failure mouse model and using a human heart tissue culture to observe Protein Z’s effect on heart function following fibrosis development will increase generalizability to humans.
A better understanding of Protein Z-mediated fibrosis may offer methods to treat fibrosis and cardiovascular illnesses. This research project furthered understanding of Protein Z’s role in cardiovascular fibrosis by demonstrating a connection between Protein Z expression and fibrosis development: downregulating Protein Z expression led to a decrease in fibroblast activity. Thus, the study may improve therapeutic options in the fight against heart disease by contributing to the understanding of heart failure and subsequent development of non-invasive treatment options. With an aging population, cardiovascular illness will continue to be the leading cause of death in the US and, consequently, require further investigation. Accordingly, this research can improve the quality of life for millions of people afflicted by heart failure.
References
[1] G. A. Mensah and D. W. Brown, “An Overview Of Cardiovascular Disease Burden In The United States,” Health Affairs, vol. 26, no. 1, pp. 38–48, Jan. 2007.
[2] Khalil, Natalie N., and Megan L. McCain. 2021. “Engineering the Cellular Microenvironment of Post-Infarct Myocardium on a Chip.” Frontiers in Cardiovascular Medicine, no. 11, 14 Jul. 2021.
[3] T. Espeland, I. G. Lunde, B. H. Amundsen, L. Gullestad, and S. Aakhus, “Myokardfibrose,” Tidsskrift for Den norske legeforening, vol. 138, no. 16 Oct. 2018.
[4] Z. Fan and J. Guan, “Antifibrotic therapies to control cardiac fibrosis,” Biomaterials Research, vol. 20, no. 1, p. 13 May 2016.
[5] Li, Zilong, Baoyu Chen, Wenhui Dong, Ming Kong, Zhiwen Fan, Liming Yu, Dongmei Wu, Jun Lu, and Yong Xu. 2019. “MKL1 Promotes Endothelial-to-Mesenchymal Transition and Liver Fibrosis by Activating TWIST1 Transcription.” Cell Death & Disease, no. 899, pp. 1–13, 19 Nov 2019.
[6] Lyons, Peter , Neil R. Mattatall, and Hyo-Sung Ro. 2006. “Modeling and Functional Analysis of AEBP1, a Transcriptional Repressor.” Proteins, no. 4, pp. 1069-1083, 1,Jun 2006.
[7] Younesi, Fereshteh S., Dong Ok Son, Joao Firmino, and Boris Hinz. 2021. “Myofibroblast Markers and Microscopy Detection Methods in Cell Culture and Histology.” Methods Mol Bio, no. 2279, pp.17–47, 25 May 2021.
[8] Roshdy, Ashraf, Shroque Zaher, Hossam Fayed, and John Gerry Coghlan. “COVID-19 and the Heart: A Systematic Review of Cardiac Autopsies.” Front Cardiovasc Med, 7 January 2021
[9] Wang, N. Rabhi, S.-F. Yet, S. R. Farmer, and M. D. Layne, “Aortic carboxypeptidase-like protein regulates vascular adventitial progenitor and fibroblast differentiation through myocardin related transcription factor A,” Sci Rep, vol. 11, no. 1, p. 3948, Feb. 2021.
[10] Elsafadi et al., “Transgelin is a TGFβ-inducible gene that regulates osteoblastic and adipogenic differentiation of human skeletal stem cells through actin cytoskeleston organization,” Cell Death Dis, vol. 7, no. 8, p. 2321, Aug. 2016.
[11] Liu et al., “AEBP1 promotes epithelial-mesenchymal transition of gastric cancer cells by activating the NF-κB pathway and predicts poor outcome of the patients,” Sci Rep, vol. 8, no. 1, Aug. 2018.
[12] R. Alpert et al., “A mechanistic analysis of the force- frequency relation in non-failing and progressively failing human myocardium,” Basic Res Cardiol, vol. 93, pp. 23–32, Mar. 1998.
[13] Keteepe-Arachi, “Cardiac Fibrosis in Hypertension”, J Hypertens Manag, vol. 3, no. 1, Feb. 2017.
[14] M. W. Schmidt and R. E. Schmieder, “Aldosterone- induced cardiac damage: focus on blood pressure independent effects,” Am J Hypertens. vol. 16, no. 1, pp. 80–86, Jan. 2003.
[15] Van der Pol A et al., “Treating oxidative stress in heart failure: past, present and future.” Eur J Heart Fail. 2019;21 no. 4, pp. 425-435, 19 Oct 2018.
[16] TS Shankar, “Understanding Cardiac Remodeling and Reverse Remodeling in Heart Failure.” Ph.D. dissertation, Department of Biomedical Engineering, University of Utah, SLC, United States, 2022
Media Attributions
- Fig1
