Mutations Which Modify Neurotransmitter Release
Dentyn Sacre
This is a first draft which has not been edited. If you have questions, or want to help in the writing or editing process, please contact the author: dentynsacre@mail.weber.edu
Genetics encode for the many different proteins involved in neurotransmitter release and binding, including receptors, their subunits, their location, the vesicles that package the neurotransmitters, and proteins involved in intracellular signaling and metabolism.
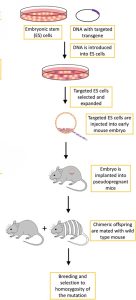
To investigate the functional roles of different genes in neurotransmission, researchers use knockout mice, which are genetically modified mice with an existing gene modified or inactivated, hence their name “knockout.” To make a gene inactive, scientists engineer a new DNA sequence after the original gene of interest, changing the sequence sufficiently to make it inoperable. They also code a marker gene into this new sequence, whose purpose will become clearer later.
The process of creating a knockout mouse starts with the isolation of embryonic stem cells from a mouse blastocyst, where the new genetic sequence is introduced to the embryonic stem cells through electroporation, a technique of applying an electric field to a cell to increase the permeability of its membrane. Then, homologous recombination is responsible for sewing the new gene into the larger chromosome. If incorporation occurs in both of the homologous chromosomes, the process was successful and the cells are said to be homozygous, but the chances of this is fairly low. It is common for these cells to come out of homologous recombination with the new gene in only one of the chromosomes, and they are said to be heterozygous. The altered cells are then separated from the unaltered ones using the marker gene and inserted into a blastocyst, which is then implanted into the uterus of a mouse with a different fur color.
The mice born will have some resulting tissues from the stem cells of the female mouse’s embryo, and others from the inserted knockout stem cells. These mice are known as chimeras due to the heterogeneity of their body parts, and their fur will have patches of different colored fur. If the original mouse the embryonic stem cells were obtained from was white and the mouse who mothered the chimeras was grey, the white patches of fur would indicate that it had come from the altered stem cells, and the grey patches from the blastocyst. A chimera is then bred with a normal mouse, and the resulting litter will contain some mice with heterozygous copies of the altered gene. Further breeding occurs until homozygous knockout mice emerge, expressing no normal form of the intended knockout gene. Because the genetic engineering occurs in stem cells, the building blocks for all subsequent cells in the organism, the resulting mouse will contain the altered sequence in all of its cells.
 https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2018.00268/full
https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2018.00268/full
CC BY 4.0
Illustration of Knockout Mice Genetic Engineering Process[1]
Research into knockout mice provide insight on primarily the function of the different neurotransmitters, receptors, proteins, and enzymes involved in neurotransmission, but they also are informative for the sake of understanding the role of different genes in individual differences of cognitive function or etiology of congenital neurological conditions.
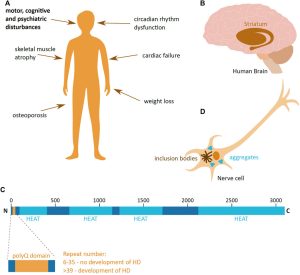
For example, Huntington’s Disease, a neurodegenerative disease with no cure resulting from a mutation in the HTT gene, has benefited greatly from animal model research. A mutant form of the HTT protein, mHTT, is produced when a mutation causing an inflation in the number of CAG repeats at the end of the gene sequence, and is cloven by enzymes into small, toxic fragments—an otherwise innocuous process in the normal HTT protein. These toxic fragments have the tendency to aggregate in neurons, leading to cell damage and cell death. Because it mostly affects neurons in the basal ganglia, the area of the brain responsible for controlling movement as well as aspects of mood, before later spreading to other parts of the brain, Huntington’s patients typically suffer hyperkinetic, jerky-like movement and psychiatric disturbances such as depression.
https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2021.769184/full
CC BY 4.0
A Visualization of Huntington’s Disease[2]
Animal models expressing Huntington’s Disease like pathologies, like R6/2 and HdhR150 mice strains genetically modified to undergo similar over repetition of the CAG segment of the HTT gene, have been instrumental in pioneering gene silencing techniques, which can be use to reduce mHTT production, and stem cell therapy aiming to replace damaged neurons.
Other uses of animal models have investigated the result of complete gene knockouts, giving us deeper insight into the function of different proteins and neurotransmitters.
Catecholamines: Dopamine and Norepinephrine
Tyrosine hydroxylase is the enzyme that catalyzes the conversion of the amino acid L-Tyrosine to L-Dopa, also known as levodopa, the precursor to dopamine. When deprived of the gene coding for this enzyme, mice are not viable.
Mice deprived of Dopamine-𝛽 hydroxylase, which catalyzes the conversion of dopamine to norepinephrine, display hypotension, sleep dysregulation, and hypersensitivity to psychostimulants.
Knockout of genes coding for monoamine oxidase, mitochondrial enzymes which catalyze oxidative degradation of amine neurotransmitters like serotonin, norepinephrine, and dopamine, causes aggressive behaviors.
Knockout of catechol-o-methyltransferase (COMT), one of several enzymes that degrade catecholamines, leads to aggression in heterozygous mice.
Homozygous vesicular monoamine transporter 2 (VMAT-2) are not viable, while heterozygous knockouts have hypersensitivity to psychostimulants. VMAT-2 is a protein that transports neurotransmitters like serotonin or dopamine into synaptic vesicles.
Dopamine transporter (DAT), a membrane spanning protein, is crucial for the reuptake of dopamine from the synaptic cleft into the cytosol. Knockouts experience growth defects, hyperlocomotion, impaired working memory, and sleep dysregulation.
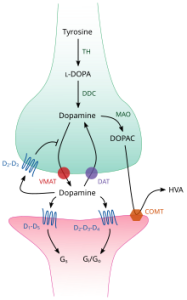
Dopamine cycle in the synapse. Dopamine release is facilitated by VMAT, and broken down with DAT.
Norepinephrine transporter (NET) is responsible for sodium-calcium dependent reuptake of extracellular norepinephrine, as well as some reuptake of dopamine. Knockout leads to psychostimulant hypersensitivity, enhanced morphine-induced analgesia, and ADHD in humans.
Tyrosine hydroxylase (TOHase) is the enzyme responsible for kicking off catecholamine synthesis by converting tyrosine to L-Dopa. Mutations of TOHase leading to tyrosine hydroxylase deficiency elicits a range of effects from deficits in movement and rigidity to severe encephalopathy in humans.
Glutamate
When the AMPA glutamate receptor, specifically the GluA2 subunit, is altered genetically to be more permeable to calcium ions, the mice will develop seizures and die by three weeks of age. This is a result of the ineditable GluA2 subunits. Normally, mRNA editing limits calcium permeability. The editing enzyme is RAD1, of which deletion is also lethal. GluA2 editing is essential for proper brain function.
Complete deletion of the GluA2 allele also increases calcium permeability, but does not lead to death or seizures. Instead, it causes a form of long-term potential in homozygous mice. Normally, long-term potentiation is driven by activation of AMPAR by sodium influx and some potassium efflux, with calcium barred from entering lest the neuron become overexcited (see the chapter on the Nernst Potential). This could be due to the genetic modification leading to disruption in AMPA assembly, reducing the number of functional receptors in the synapse. Less receptors, even if they are more permeable to calcium, could remain well below the excitotoxic threshold.
Mutations targeting the NMDA GluN1 subunit interferes with breathing and is lethal in only a few hours after birth. When the mutation is concentrated to only the CA1 region of the hippocampus, mice grow normally but exhibit memory difficulty in spatial maze tasks, suggesting the CA1 region of the hippocampus is closely tied to spatial memory.
Another strain of mice called “Doogie mice” or “smart mice,” engineered to overexpress the NR2B gene which codes for a protein subunit of NMDA, show enhancements in learning and memory. Prolonged NMDAR channel opening leads to improvements in coincidence detection, long-term potentiation, and memory, with sustained cognitive benefits in old age. This gain of function mutation supercharges plasticity and contrasts the function of NR2B with NR2A, the subunit with a shorter opening time that is dominant in adult cortex and hippocampus. NR2A has been associated with less calcium influx and more cognitive stability.
Gain of function: genetic engineering leading to an enhancement of function rather than a deficit of function.
Mice lacking a functional mGluR1 gene, coding for the first of glutamate’s metabotropic receptors, develop dysfunction in the cerebellum, expressed in an ataxic gait, tremor, and dysmetria. In mGluR2 knockout mice, the gene coding for the second metabotropic glutamate receptor, presynaptic inhibition at mossy fiber synapses was reduced, indicating mGluR2 plays a role in presynaptic regulation of neurotransmission. This did not affect long-term potentiation in the mice, but did affect long-term depression significantly. Knockout of mGluR4 lead to impairments in presynaptic inhibition at parallel fiber PC synapses, reducing efficiency of synapses after repetitive firing. mGluR8 is the exclusive mediator of presynaptic inhibition and the lateral perforant pathway in mice.
Vesicles
Neurotransmitters travel via vesicular transportation. Mutations in the genes coding for vesicle formation could lead to downstream effects in transmission. For example, there has been some evidence that the SNAP-25 gene, involved in the process of neurotransmitter release, is related to ADHD. Researchers have shown that a deletion of the gene in animal models causes hyperactivity similar to that seen in ADHD[3].
Prion Disease
Prions are infectious, misfolded proteins which are almost exclusively forms of PrP, the protein coded for by the PRNP gene whose function is largely unknown, although it’s healthy form, PrPC is suspected to regulate copper or zinc metabolism. PrPSC, the disease causing form of the protein resulting from, can be caused by spontaneous misfolding, mutations in the PRNP gene, or ingestion of diseased tissue containing PrPSC, such as infected, mad cow disease causing meat. This infectious protein aggregates into plaques inside of neurons, blocking normal function.
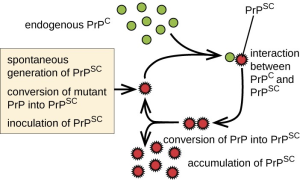
Mice completely lacking PRNP survive and are resistant to prions, however deletions beyond residue 106 of the gene causes ataxia and cerebellar granule loss. Other proteins, such as the Shadoo protein, may compensate for the loss of PrP protein in PRNP knockouts. These mice show no major lifespan or fertility issues besides some altered synaptic physiology and changes in circadian rhythm, cognition, and olfaction.
Peptides
Carboxypeptidase E (CPE) is an enzyme involved in the synthesis of most neuropeptides and peptide hormones and is controlled by the CPE gene. CPE knockout mice are viable, but secrete proinsulin rather than insulin, leading to diabetes.
Knockout of Enkephalin, the peptide involved in the regulation of pain, leaves mice healthy enough to reach adulthood. They display a normal tail-flick response, indicating no role of enkephalin in baseline pain, but lose centrally mediated analgesia. Exploratory activity is diminished in these mice and aggression is more common. On the other hand, mice engineered to under-express NK1R, the receptor that substance P binds to in creating the sensation of pain, reduces pain sensitivity.
Axonal Growth
Neurite outgrowth inhibitor, also known as reticulon 4 or Nogo, is a protein coded for by the RTN4 gene responsible for inhibiting neurite growth in the adult mammalian CNS. It exists in three isoforms: Nogo-A, Nogo-B, and Nogo-C. Blocking Nogo A promotes axonal regeneration and functional recovery in spinal-cord injuries. Blocking all three isoforms is often lethal.
- Lampreht, T. U., Horvat, S., & Cemazar, M. (2018). Transgenic mouse models in cancer research. Frontiers in Oncology, 8:268. https://doi.org/10.3389/fonc.2018.00268 ↵
- Jarosinska, O. D., & Rudiger, S. G. D. (2021). Molecular strategies to target protein aggregation in Huntington's Disease. Frontiers in Molecular Biosciences, 8:769184. doi:10.3389/fmolb.2021.769184 ↵
- Brophy, K., Hawi, Z., Kirley, A., Fitzgerald, M., & GIll, M. (2002). Synaptosomal-associated protein 25 (SNAP-25) and attention deficit hyperactivity disorder (ADHD); Evidence of linkage and association in the Irish population. Molecular Psychiatry, 7(8), 913-917. https://doi.org/10.1038/sj.mp.4001092 ↵
