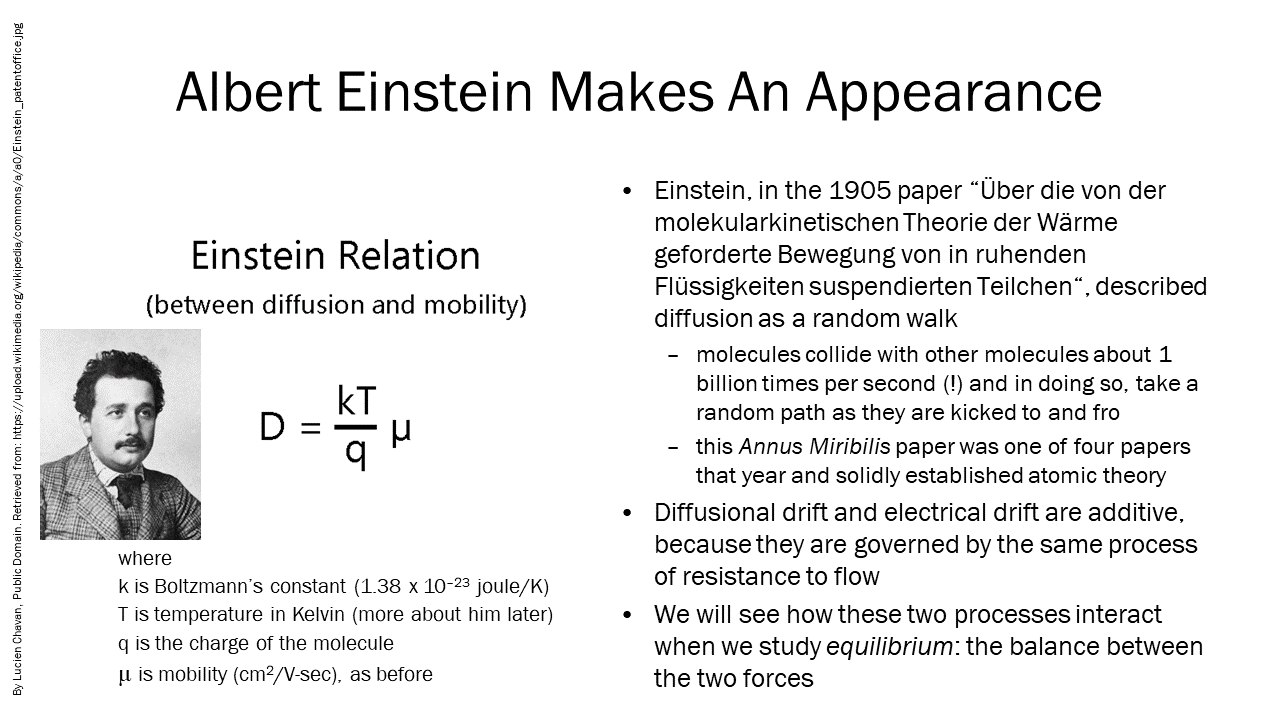Lennox-Gastaut Syndrome
Kobe Christensen and Jim Hutchins
LENNOX-GASTAUT SYNDROME
Introduction
Lennox-Gastaut (LGS) is an uncommon pediatric epilepsy syndrome that makes up around 10% of childhood epilepsy. Lennox-Gastaut is characterized by the onset of multiple different seizure types, severe cognitive delay, and a distinct EEG pattern.
History
Lennox-Gastaut is named after the two physicians who first documented the disease. In the 1950’s, Dr. William Lennox described a particular EEG finding that would match an epilepsy syndrome that Dr. Henri Gastaut was studying in the mid 1960’s. Dr. Henri Gastaut described a childhood epilepsy with frequent tonic and absence seizures. Later it was found that both Dr. Lennox and Dr. Gastaut were studying the same epilepsy syndrome and thus it was coined Lennox-Gastaut syndrome.
Symptoms
Lennox-Gastaut is frequently associated with intractable epilepsy, severe cognitive impairment, and characteristic EEG.
Epilepsy
Patients with Lennox-Gastaut tend to present with multiple seizure types.
- Tonic
- Tonic-Clonic
- Atonic (Drop attacks)
- Myoclonic
- Atypical Absence (With characteristic EEG pattern)
Seizures in patients with Lennox-Gastaut are frequent in nature and tend to cluster making them at high risk for evolving into Non-convulsive status epilepticus (NCSE) with some studies showing an incidence of NCSE being over.
The average of seizure onset in patients with LGS is between 3-5 years of age. Almost all patients with an LGS diagnosis have an onset of seizures before 8 years of age.
Cognitive Impairment
Cognitive impairment before seizure onset is seen in roughly half of patients. Depending on the severity of the disease there may be a delayed onset of cognitive delay after the patient has their first seizure.
Characteristic EEG pattern
EEG in patients with LGS can be complex and difficult to read. Due to the multitude of seizure types, LGS seizure can present in several different ways which may make it difficult to differentiate the disease from others. However, in LGS there is a very characteristic EEG pattern known as atypical absence which helps to set it apart from others.
Atypical absence seizures are defined as slow (1.5-2.5 Hz) irregular polyspike and wave activity. It is important to note that atypical absence seizures differ from typical absence seizures in that they are slower (Typical absence seizures tend to be in the 3-5 Hz range) and are often asymmetric (Typical absence seizures are almost always generalized). Atypical absence seizures are NOT triggered by hyperventilation.
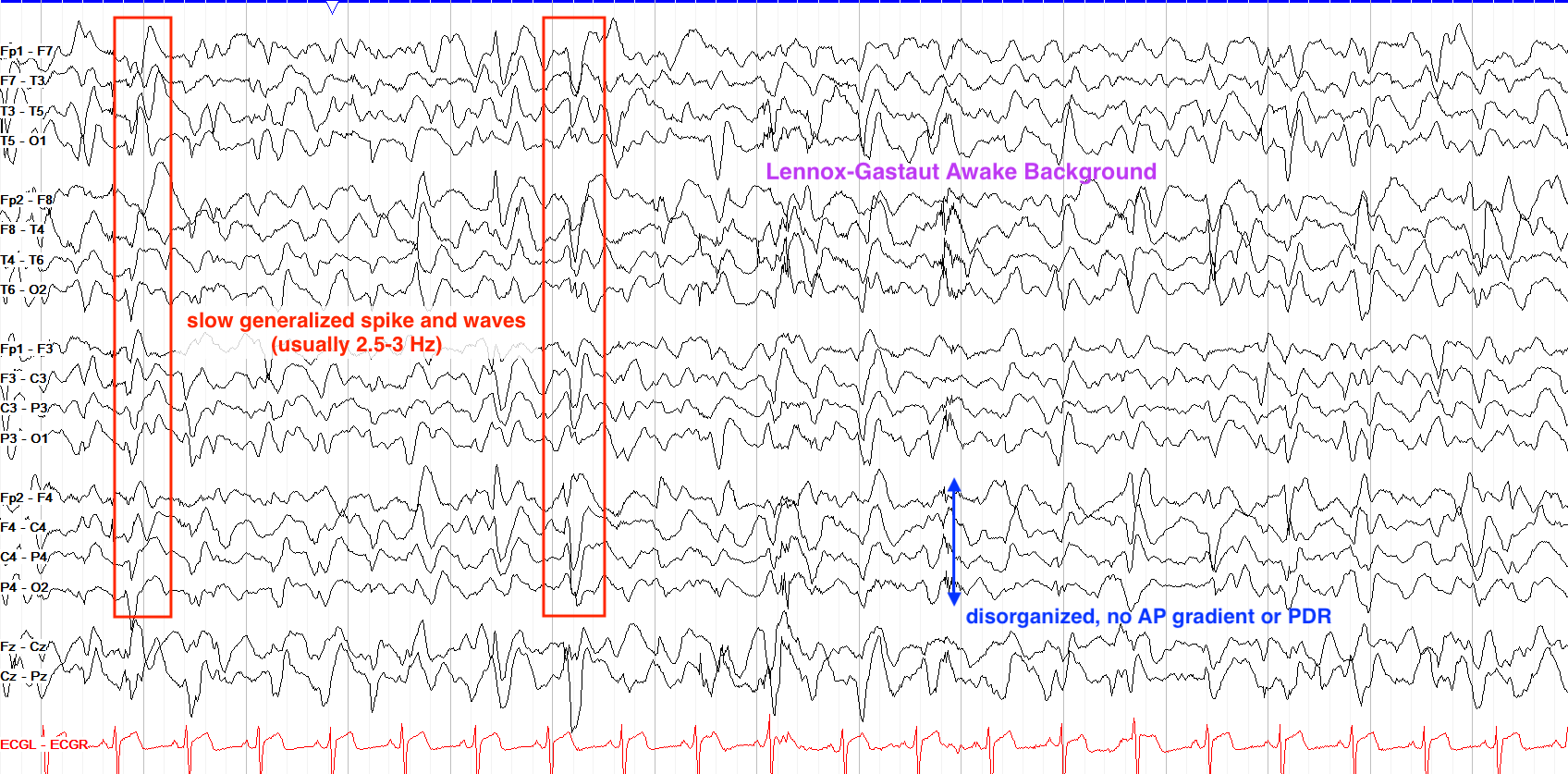
From “The Pediatric EEG”, by David Valentine M.D., 2020, (https://www.learningeeg.com/pediatric). Copyright 2020 by David Valentine
Figure 2.1 showing Lennox Gastaut awake background with 2-2.5 Hz generalized spike and wave.
Other patterns that can be seen in Ictal EEG of an LGS patient include:
- Rhythmic, high amplitude discharges in the high alpha-beta ranges seen in tonic seizures.
- Arrhythmic bursts of polyspike-wave discharges seen in myoclonic seizures.
Classifications of disease
LGS diagnoses typically fall into the following two categories: Secondary & Idiopathic
Secondary LGS:
This accounts for about 75% of all cases and describes a known primary cause. Any damage to the brain before or during birth can be considered a primary cause to LGS. These include:
- Infection
- Frontal lobe injury
- Tuberous Sclerosis
- Perinatal asphyxia
- Perinatal stroke
- Abnormal development
Idiopathic LGS
These account for the other 25% of LGS cases and have no known cause.
Linkage between Lennox-Gastaut & West Syndrome
An LGS diagnosis has been to known to frequently follow a myriad of other childhood epilepsy diagnoses. None are more common than the diagnosis of West Syndrome, as many LGS cases follow a diagnosis of West Syndrome despite no known cause for the linkage of the two.
Treatments & Prognosis
Long-term prognosis is generally unfavorable for patients with LGS. While mortality secondary to LGS is low, severe cognitive impairment and treatment resistant epilepsy still persists in the majority of patients. Patients with a history of West Syndrome also tend to have worse outcomes than those with idiopathic LGS.
Treatment options in LGS vary due to the multitude of seizure types. Antiepileptic drugs (AEDs) tend to be the mainline treatment for LGS but rarely does a single AED give complete relief of seizures. Valproic Acid (Depakote) and Lamotrigine (Lamictal) are the most commonly used AEDs in LGS. Other forms of treatment include the Ketogenic Diet, VNS, and surgical intervention. LGS patients are good candidates for corpus callostomy to help reduce the frequency of seizures, though it is rare for surgical intervention to be completely curative.
Key Takeaways
- 1.5-2.5 Hz Absence seizures are a common EEG finding in patients with LGS.
- LGS is likely to be secondary to some form of neurological damage before or during birth.
- There is a common progression of West Syndrome to Lennox-Gastaut.
- Valproic acid is the mainline treatment for LGS, followed by a variety of other AEDs as needed.
MYOCLONIC-ASTATIC EPILEPSY (DOOSE SYNDROME)
Introduction
Myoclonic-Astatic Epilepsy (MAE), also known as Doose Syndrome is a pediatric seizure disorder commonly affecting children between the ages of 2-5 years that is characterized by frequent myoclonic and atonic seizures. MAE often presents with developmental delay, cognitive impairment, and behavioral challenges.
MAE affects males more often than females at a ratio of 2:1 after the first year of life. During the first year of life literature suggests that MAE will affect the sexes at roughly the same rate.
Signs & Symptoms
Epilepsy
MAE's hallmark symptoms are frequent myoclonic and atonic seizures, however it is not uncommon for other seizure types to be witnessed in patients with MAE. Other seizures may include generalized tonic-clonic, absence, tonic, and progression to non-convulsive status epilepticus (NCSE). Patients can have hundreds of seizures in a single day, and most frequently have seizures in the morning shortly after waking. Sometimes these seizures are isolated, but they often tend to cluster thus increasing their risk to slip into status-epilepticus.
EEG in MAE typically reveals bursts of 2-5 Hz polyspike and wave complexes that are generally superimposed onto otherwise normal looking backgrounds. An important feature in differentiating MAE from other pediatric seizure disorders like LGS is the presence of a relatively normal PDR and sleep architecture.
Cognitive Impairment
Cognitive outcomes in MAE are highly variable. Some experience normal cognition while others may have severe intellectual disability. The literature suggests that seizure frequency is correlated to the cognitive outcome as those who attain freedom from seizures tend to have more favorable cognitive outcomes. Changes in behavior are another commonly associated symptom with MAE. Up to 1/5th of documented MAE cases are diagnosed with ADHD.
Treatment & Prognosis
Treatment efficacy of MAE varies as some enter complete remission of seizures, and others have intractable epilepsy. MAE is largely considered a generalized seizure disorder and is treated as such with ethosuximide which is a common AED for controlling generalized seizures. The standard of care is to pair the AED's with dietary therapy which most often consists of the ketogenic diet.
Prognosis in MAE varies depending on the age of onset, frequency of seizures, and seizure control. The literature suggests that roughly 3 in 5 children will have a complete remission from seizures and no longer require treatment. In other children, seizure control may range from well controlled to intractable. In children who do not have seizure freedom, cognitive impairment tends to increase in severity.
Key Takeaways
- Myoclonic-Astatic Epilepsy (MAE) is characterized by frequent myoclonic and atonic seizures.
- Bursts of 2-5 Hz polyspike and wave discharges superimposed onto otherwise normal backgrounds are a common characteristic of MAE.
- Prognosis is often dependent on factors such as seizure frequency, age of onset, and seizure type.
- Dietary therapy is effective in the treatment of MAE.
RETT SYNDROME
Introduction
Rett Syndrome is a rare pediatric neurodevelopmental disorder that primarily affects females and is characterized by severe cognitive and physical impairment. Rett Syndrome is primarily caused by the MECP2 gene, which plays a crucial role in regulating gene expression during brain development.
Symptoms
Cognitive Impairment
The hallmark symptom of Rett Syndrome is a loss of purposeful gross motor skills. Rett Syndrome has a slow onset of symptoms, and is typically diagnosed between 1-2 years of age. Most commonly, parents will notice a regression of acquired skills like language and social engagement followed by a regression in large motor skills (I.E. Patient goes from walking to primarily crawling). Along with a loss of purposeful motor movement, repetitive and uncontrolled hand movements such as hand-wringing or hand-washing motions become ever apparent in patients with Rett Syndrome. Epilepsy, scoliosis, and gait abnormalities are also common symptoms seen in Rett Syndrome.
Epilepsy
In the literature, there are not many findings of characteristic EEG patterns in patients with Rett Syndrome. Early in Rett Syndrome, EEG's typically tend to be normal. As Rett Syndrome begins to progress centrotemporal spikes may be seen with a clinical correlation of motor impairment. As Rett Syndrome further progresses abnormalities during sleep may be seen followed by diffuse background slowing and multifocal spike and wave activity.

Figure 5.1 Rett Syndrome patient with Scoliosis
Konstantinos C Soultanis, corresponding author1 Alexandros H Payatakes,2,3 Vasilios T Chouliaras,2 Georgios C Mandellos,2 Nikolaos E Pyrovolou, Fani M Pliarchopoulou,4 and Panayotis N Soucacos1, CC BY 2.5 via Wikimedia Commons
Treatment & Prognosis
Prognosis
While most individuals with Rett Syndrome live well into adulthood, cognitive prognosis remains quite poor. Almost all patients with Rett Syndrome require 24 hour support as they maintain a cognitive level to that of a 1 year old.
Treatment
There are no direct pharmacological treatments for Rett Syndrome. However, anti-epileptic drugs are frequently used to decrease seizure frequency. Various types of therapy can help to improve the quality of life for those with Rett Syndrome, but at this time there is no treatment for the cognitive impairment experienced by these patients.
Key Takeaways
- Rett Syndrome is a neurodevelopmental disorder that primarily affects females.
- Caused by a mutation in the MECP2 gene, children with Rett Syndrome tend to develop normally until 6-18 months of age.
- The hallmark symptom for Rett Syndrome is the regression of gross motor function.
- There are no known treatments or cures for Rett Syndrome.
[latex]J_{drift} = \delta_{el} E \\ J_{drift} = -\mu z [ion] \frac{\delta V}{\delta x}[/latex]

[latex]D = \frac{k T}{q}\mu[/latex]