Huntington’s Disease
Brooke Hildt and Jim Hutchins
Overview
Huntington’s Disease is an autosomal dominant trinucleotide repeat disorder that causes the progressive degeneration of the basal nuclei leading to clinical symptoms affecting voluntary movement, cognitive impairment, and psychiatric disorders.
Genetics of Huntington’s Disease
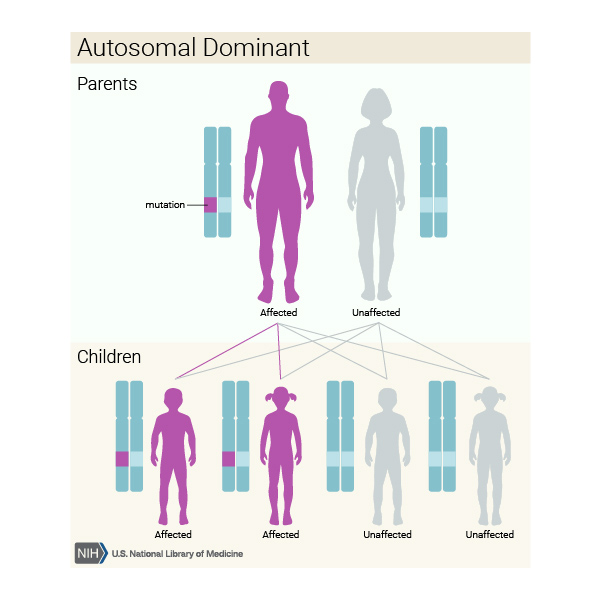
In Mendelian terms, Huntington Disease is inherited as an autosomal dominant disorder. This means, if a parent has the disease, offspring will have a 50% (1 in 2) chance of having the disease as well.
Trinucleotide Repeat Diseases
The Huntington Disease repeats are found within the exon of the huntingtin gene which is located in the short arm (p arm) of chromosome four. These repeats consist of the CAG codon, which codes for the amino acid glutamine (Gln or Q). The polyglutamine region of the expanded protein can interfere with neuronal survival. The more repeats you have, the worse the disease is, and therefore the earlier the onset of symptoms. Clinical signs typically appear around age 20-65 years due to the accumulation of mutated Huntingtin reaching toxic levels; if the individual has complete penetrance (greater than 36 CAG repeats). The CAG trinucleotide repeats expand with each generation.
Neural Wiring and Chemistry in Huntington’s Disease
X
Degeneration Process
Degeneration occurs due to the accumulation of the protein Huntingtin that destroys the medium spiny neurons located in the striatum. The striatum, known as the brain’s ”input” center, receives input from four key pathways: the motor, oculomotor, association, and limbic loops. Within each loop, the striatum functions with an inhibitory action mediated by gamma-aminobutyric acid (GABA). In Huntington’s disease, an imbalance between inhibitory and excitatory signals in these pathways leads to symptoms such as involuntary movements, difficulty controlling eye movements, impaired cognitive abilities like planning and directing movement, and problems with regulating emotions and motivation related movement.
Signs and Symptoms of Huntington’s Disease
Movement disorders
- Trouble with gait, posture, or maintaining balance
- Muscle rigidity or contracture
- Slow or unusual eye movements
- Trouble with speech or swallowing
- Involuntary muscle contractions or spasms
Cognitive conditions
- Lack of awareness
- Anterograde amnesia
- Trouble organizing, prioritizing, or focusing on tasks
- Lack of impulse control
- Perseveration
Mental heath conditions
- Depression
- Trouble sleeping
- Fatigue and loss or energy
- Social withdrawal
- Irritability
Diagnosis
A preliminary diagnosis of Huntington’s disease generally includes a neurological examination, neurophysiological testing, a mental health evaluation, brain imaging and function tests, and genetic testing.
Treatment
Huntington’s disease is unfortunately a fatal condition with no cure. After onset of clinical symptoms, the patient will have 10-20 years of remaining longevity. Symptomatic management is best practice.
Case
CC BY-SA adapted from Physio-Pedia
45-year-old Caucasian male presented with an increase in accidents at work over the last couple months: accidently cutting fingers, dropping plates, etc. ER physician noticed the patient was stumbling often and had difficulty with upper limb coordination; also noted pt seemed to have a short temper. Patient was referred to a neurologist. The patient had a past medical history of hypertension x5 years. No relevant family history. Objective findings revealed patient had unremarkable vital signs, was aware and oriented, exhibited normal posture, had a slight hesitant gait, some difficulty with cognitive functioning testing, occasional repeated involuntary movements of both upper and lower extremities and facial twitching, and a manual muscle test revealing concerning outcomes. The patient’s medical diagnosis disclosed early-stage Huntington’s Disease due to a lesion to the basal ganglia. Interventions would include physiotherapy, occupational therapy, and motivation therapies. Short and long term goals were set to keep the patient motived. Short term goals: perform balance related training activated 5x per week, learn effective cutting safety techniques, perform 30 min of aerobic exercise training 4x days per week. Long term goals: participate in an organized skating session once per week, continue to ambulate 500m without gait aid, walk dog 2x a week.
Media Attributions
- GHR_Inheritance_Images © National Library of Medicine is licensed under a Public Domain license

{kind=link}